学习最好的方式就是分享。
最近看到一个在R上进行的RNA-seq 分析流程,恰好自己也有过RNA-seq分析的经验,所以就想结合以前的经验分享这个流程出来。P.S. RNA-seq 分析有多种流程,本文仅是举出其中一个例子,抛砖引玉。
本文将要介绍的是由Combine Australia 所提供的一个针对有参基因组的基因差异表达分析流程。
分析流程的基本信息
目标物种 : mouse (小鼠)
组织 : mammary gland (乳腺)
生理状态:
virgin (未怀孕)
pregnant (怀孕)
lactating (产后哺乳期)
细胞类型:
basal stem-cell enriched cells (B)
committed luminal cells (L)
Six groups (3 conditions x 2 cell types) with 2 biological replicates per group
该文章已经发表在Nature cell biology :
流程
数据 感兴趣的朋友也可以下载本次分析所需要的数据集,跟着动手尝试:https://figshare.com/s/1d788fd384d33e913a2a
配置
安装了R语言(3.5.0以上)及R studio(可选)
需要的R包: limma, edgeR, gplots, org.Mm.eg.db, EDASeq, RColorBrewer, GO.db, BiasedUrn, DESeq2, Glimma, Rsubread.
这里我们推荐使用Bioconductor安装以上这些包
1 BiocManager::install(c ('limma' , 'edgeR' , 'gplots' , 'org.Mm.eg.db' , 'EDASeq' , 'RColorBrewer' , 'GO.db' , 'BiasedUrn' , 'DESeq2' , 'Rsubread' ))
这里要注意Glimma包需要通过在Bioconductor 中下载其源文件来安装
源文件的安装方法之一 源文件可以在R studio中安装,在上方工具栏中选择**”Tools –> Install packages”**。
然后,在弹出的窗口中点击**”Install from”下拉菜单中选择 Package Archive File**。
最后,选择下载的源文件包即可安装。
Step 2 RNA-seq Pre-processing
Ref: https://bioinformatics-core-shared-training.github.io/RNAseq-R/rna-seq-preprocessing.nb.html
本节将要介绍的是在R中进行RNA-seq数据预处理的实战代码
主要包括以下方面:
载入mapping, counting后的数据
过滤低表达基因
对表达数据进行质量控制
标准化处理
本次流程需要的包
1 2 3 4 5 6 library(edgeR) library(limma) library(Glimma) library(gplots) library(org.Mm.eg.db) library(RColorBrewer)
温馨提示:org.Mm.eg.db有60多M,个人推荐没有安装过的朋友先切换到国内的镜像,再下载这个包。
1 2 3 options(repos = 'https://mirrors.tuna.tsinghua.edu.cn/CRAN/') getOption('repos') BiocManager::install('org.Mm.eg.db')
另外,本次所需要的数据均可以在该网站下载:https://figshare.com/s/1d788fd384d33e913a2a
下载的数据有:
Sampleinfo.txt
GSE60450_Lactation-GenewiseCounts.txt
mouse_c2_v5.rdata
mouse_H_v5.rdata
载入数据 首先,我们读入样本信息**”SampleInfo.txt”**
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 > sampleinfo <- read.delim("data/SampleInfo.txt" ) > sampleinfo FileName SampleName CellType Status 1 MCL1.DG_BC2CTUACXX_ACTTGA_L002_R1 MCL1.DG luminal virgin2 MCL1.DH_BC2CTUACXX_CAGATC_L002_R1 MCL1.DH basal virgin3 MCL1.DI_BC2CTUACXX_ACAGTG_L002_R1 MCL1.DI basal pregnant4 MCL1.DJ_BC2CTUACXX_CGATGT_L002_R1 MCL1.DJ basal pregnant5 MCL1.DK_BC2CTUACXX_TTAGGC_L002_R1 MCL1.DK basal lactate6 MCL1.DL_BC2CTUACXX_ATCACG_L002_R1 MCL1.DL basal lactate7 MCL1.LA_BC2CTUACXX_GATCAG_L001_R1 MCL1.LA basal virgin8 MCL1.LB_BC2CTUACXX_TGACCA_L001_R1 MCL1.LB luminal virgin9 MCL1.LC_BC2CTUACXX_GCCAAT_L001_R1 MCL1.LC luminal pregnant10 MCL1.LD_BC2CTUACXX_GGCTAC_L001_R1 MCL1.LD luminal pregnant11 MCL1.LE_BC2CTUACXX_TAGCTT_L001_R1 MCL1.LE luminal lactate12 MCL1.LF_BC2CTUACXX_CTTGTA_L001_R1 MCL1.LF luminal lactate
再读入基因计数后的数据**”GSE60450_Lactation-GenewiseCounts.txt”**
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 > seqdata <- read.delim("data/GSE60450_Lactation-GenewiseCounts.txt" , + stringsAsFactors = FALSE ) > head(seqdata) EntrezGeneID Length MCL1.DG_BC2CTUACXX_ACTTGA_L002_R1 MCL1.DH_BC2CTUACXX_CAGATC_L002_R1 1 497097 3634 438 300 2 100503874 3259 1 0 3 100038431 1634 0 0 4 19888 9747 1 1 5 20671 3130 106 182 6 27395 4203 309 234 MCL1.DI_BC2CTUACXX_ACAGTG_L002_R1 MCL1.DJ_BC2CTUACXX_CGATGT_L002_R1 1 65 237 2 1 1 3 0 0 4 0 0 5 82 105 6 337 300 MCL1.DK_BC2CTUACXX_TTAGGC_L002_R1 MCL1.DL_BC2CTUACXX_ATCACG_L002_R1 1 354 287 2 0 4 3 0 0 4 0 0 5 43 82 6 290 270 MCL1.LA_BC2CTUACXX_GATCAG_L001_R1 MCL1.LB_BC2CTUACXX_TGACCA_L001_R1 1 0 0 2 0 0 3 0 0 4 10 3 5 16 25 6 560 464 MCL1.LC_BC2CTUACXX_GCCAAT_L001_R1 MCL1.LD_BC2CTUACXX_GGCTAC_L001_R1 1 0 0 2 0 0 3 0 0 4 10 2 5 18 8 6 489 328 MCL1.LE_BC2CTUACXX_TAGCTT_L001_R1 MCL1.LF_BC2CTUACXX_CTTGTA_L001_R1 1 0 0 2 0 0 3 0 0 4 0 0 5 3 10 6 307 342 > dim (seqdata) [1 ] 27179 14
由于我们关注的是基因计数的信息,因此为了方便下游分析处理,我们移除seqdata中前两列,并将第一列的ID命名为行名
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 > countdata <- seqdata[,-(1 :2 )] > rownames(countdata) <- seqdata[,1 ] > head(countdata) MCL1.DG_BC2CTUACXX_ACTTGA_L002_R1 MCL1.DH_BC2CTUACXX_CAGATC_L002_R1 497097 438 300 100503874 1 0 100038431 0 0 19888 1 1 20671 106 182 27395 309 234 MCL1.DI_BC2CTUACXX_ACAGTG_L002_R1 MCL1.DJ_BC2CTUACXX_CGATGT_L002_R1 497097 65 237 100503874 1 1 100038431 0 0 19888 0 0 20671 82 105 27395 337 300 MCL1.DK_BC2CTUACXX_TTAGGC_L002_R1 MCL1.DL_BC2CTUACXX_ATCACG_L002_R1 497097 354 287 100503874 0 4 100038431 0 0 19888 0 0 20671 43 82 27395 290 270 MCL1.LA_BC2CTUACXX_GATCAG_L001_R1 MCL1.LB_BC2CTUACXX_TGACCA_L001_R1 497097 0 0 100503874 0 0 100038431 0 0 19888 10 3 20671 16 25 27395 560 464 MCL1.LC_BC2CTUACXX_GCCAAT_L001_R1 MCL1.LD_BC2CTUACXX_GGCTAC_L001_R1 497097 0 0 100503874 0 0 100038431 0 0 19888 10 2 20671 18 8 27395 489 328 MCL1.LE_BC2CTUACXX_TAGCTT_L001_R1 MCL1.LF_BC2CTUACXX_CTTGTA_L001_R1 497097 0 0 100503874 0 0 100038431 0 0 19888 0 0 20671 3 10 27395 307 342
另外,我们注意到countdata中的列名过于冗长,而样本信息中的名字也只是前7位字母而已,因此我们使用substr()函数截取列名
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 > sampleinfo$SampleName [1 ] MCL1.DG MCL1.DH MCL1.DI MCL1.DJ MCL1.DK MCL1.DL MCL1.LA MCL1.LB MCL1.LC MCL1.LD MCL1.LE MCL1.LF 12 Levels: MCL1.DG MCL1.DH MCL1.DI MCL1.DJ MCL1.DK MCL1.DL MCL1.LA MCL1.LB MCL1.LC ... MCL1.LF> colnames(countdata) [1 ] "MCL1.DG_BC2CTUACXX_ACTTGA_L002_R1" "MCL1.DH_BC2CTUACXX_CAGATC_L002_R1" [3 ] "MCL1.DI_BC2CTUACXX_ACAGTG_L002_R1" "MCL1.DJ_BC2CTUACXX_CGATGT_L002_R1" [5 ] "MCL1.DK_BC2CTUACXX_TTAGGC_L002_R1" "MCL1.DL_BC2CTUACXX_ATCACG_L002_R1" [7 ] "MCL1.LA_BC2CTUACXX_GATCAG_L001_R1" "MCL1.LB_BC2CTUACXX_TGACCA_L001_R1" [9 ] "MCL1.LC_BC2CTUACXX_GCCAAT_L001_R1" "MCL1.LD_BC2CTUACXX_GGCTAC_L001_R1" [11 ] "MCL1.LE_BC2CTUACXX_TAGCTT_L001_R1" "MCL1.LF_BC2CTUACXX_CTTGTA_L001_R1" > colnames(countdata) <- substr(colnames(countdata), 1 , 7 ) > colnames(countdata) [1 ] "MCL1.DG" "MCL1.DH" "MCL1.DI" "MCL1.DJ" "MCL1.DK" "MCL1.DL" "MCL1.LA" "MCL1.LB" "MCL1.LC" [10 ] "MCL1.LD" "MCL1.LE" "MCL1.LF" > table(colnames(countdata)==sampleinfo$SampleName) TRUE 12
数据过滤 对于表达量过低的基因而言,其数据在差异分析中的可信度是较差的。因此,在本流程中我们先使用edgeR的cpm()函数,将counting后的数据转换为CPM(counts-per-million),并取CPM在两个重复样本中均大于0.5的基因进入后续分析。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 > myCPM <- cpm(countdata) > head(myCPM) MCL1.DG MCL1.DH MCL1.DI MCL1.DJ MCL1.DK MCL1.DL MCL1.LA 497097 18.85684388 13.77543859 2.69700983 10.45648006 16.442685 14.3389690 0.0000000 100503874 0.04305215 0.00000000 0.04149246 0.04412017 0.000000 0.1998463 0.0000000 100038431 0.00000000 0.00000000 0.00000000 0.00000000 0.000000 0.0000000 0.0000000 19888 0.04305215 0.04591813 0.00000000 0.00000000 0.000000 0.0000000 0.4903857 20671 4.56352843 8.35709941 3.40238163 4.63261775 1.997275 4.0968483 0.7846171 27395 13.30311589 10.74484210 13.98295863 13.23605071 13.469996 13.4896224 27.4615975 MCL1.LB MCL1.LC MCL1.LD MCL1.LE MCL1.LF 497097 0.0000000 0.0000000 0.00000000 0.0000000 0.0000000 100503874 0.0000000 0.0000000 0.00000000 0.0000000 0.0000000 100038431 0.0000000 0.0000000 0.00000000 0.0000000 0.0000000 19888 0.1381969 0.4496078 0.09095771 0.0000000 0.0000000 20671 1.1516411 0.8092940 0.36383085 0.1213404 0.4055595 27395 21.3744588 21.9858214 14.91706476 12.4171715 13.8701357 > thresh <- myCPM > 0.5 > head(thresh) MCL1.DG MCL1.DH MCL1.DI MCL1.DJ MCL1.DK MCL1.DL MCL1.LA MCL1.LB MCL1.LC MCL1.LD MCL1.LE 497097 TRUE TRUE TRUE TRUE TRUE TRUE FALSE FALSE FALSE FALSE FALSE 100503874 FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE 100038431 FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE 19888 FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE 20671 TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE FALSE FALSE 27395 TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE MCL1.LF 497097 FALSE 100503874 FALSE 100038431 FALSE 19888 FALSE 20671 FALSE 27395 TRUE
用table()函数每个基因(每行)在几个样本中是符合条件的
1 2 3 4 5 > table(rowSums(thresh)) 0 1 2 3 4 5 6 7 8 9 10 11 12 10857 518 544 307 346 307 652 323 547 343 579 423 11433
随后,保留至少在两个样本符合条件的基因。
1 2 3 4 5 6 7 8 9 > keep <- rowSums(thresh) >= 2 > counts.keep <- countdata[keep,] > summary(keep) Mode FALSE TRUE logical 11375 15804 > dim (counts.keep) [1 ] 15804 12
我们保留了15804个相对表达可信的基因。至于,counts threshold的选择可以参考原文:
As a general rule, a good threshold can be chosen by identifying the CPM that corresponds to a count of 10, which in this case is about 0.5. You should filter with CPMs rather than filtering on the counts directly, as the latter does not account for differences in library sizes between samples.
创建表达矩阵 接着我们使用edgeR的DGEList函数来创建表达矩阵
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 > dgeObj <- DGEList(counts.keep) > dgeObj An object of class "DGEList" $counts MCL1.DG MCL1.DH MCL1.DI MCL1.DJ MCL1.DK MCL1.DL MCL1.LA MCL1.LB MCL1.LC MCL1.LD MCL1.LE 497097 438 300 65 237 354 287 0 0 0 0 0 20671 106 182 82 105 43 82 16 25 18 8 3 27395 309 234 337 300 290 270 560 464 489 328 307 18777 652 515 948 935 928 791 826 862 668 646 544 21399 1604 1495 1721 1317 1159 1066 1334 1258 1068 926 508 MCL1.LF 497097 0 20671 10 27395 342 18777 581 21399 500 15799 more rows ...$samples group lib.size norm.factors MCL1.DG 1 23218026 1 MCL1.DH 1 21768136 1 MCL1.DI 1 24091588 1 MCL1.DJ 1 22656713 1 MCL1.DK 1 21522033 1 7 more rows ... > names (dgeObj) [1 ] "counts" "samples"
根据CellType和小鼠的Status,我们可以创建分组信息。
1 2 group <- paste(sampleinfo$CellType,sampleinfo$Status,sep="." ) group <- factor(group)
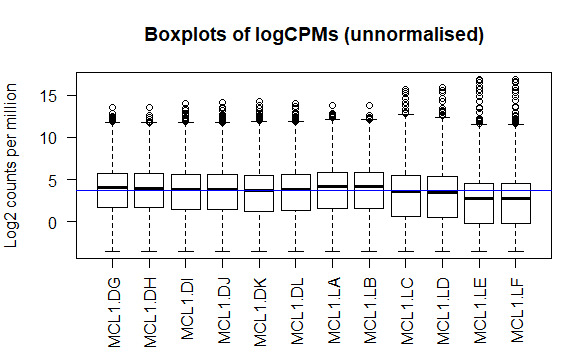
质量控制 由于我们的数据并不是正态分布的,为了下游分析的进行,使用cpm函数对我们的数据取log2处理
1 2 3 4 5 6 > logcounts <- cpm(dgeObj,log =TRUE ) > > boxplot(logcounts, xlab="" , ylab="Log2 counts per million" ,las=2 ) > > abline(h=median(logcounts),col="blue" ) > title("Boxplots of logCPMs (unnormalised)" )
从箱线图中可以看出样本数据分布虽然存在差异,但这种差异程度是我们还能接受的程度。
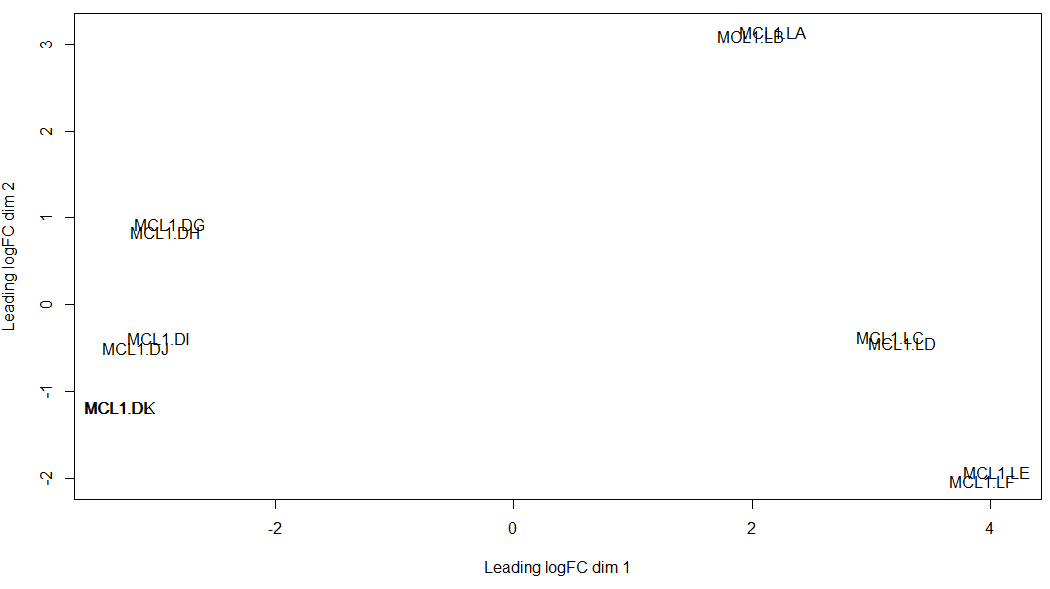
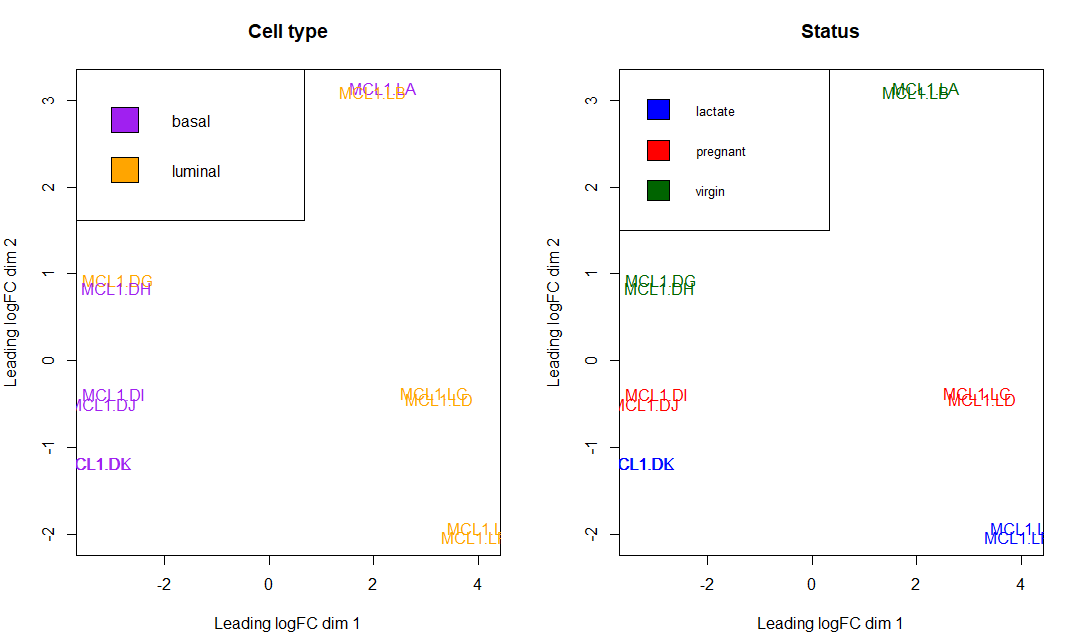
主成分分析 主成分分析在RNA-seq分析中是一个较为重要的步骤,其指出了样本数据中造成主要差异的因子是什么。一般而言,我们当然希望数据中最大的差异是由于处理与否 引起的
可以看到两个生物学重复样本都能较好的聚在一起
另外,我们也可以运用各种参数使主成分分析的可视化更加符合我们的要求,例如根据细胞类型或小鼠状态标注颜色:
总而言之,利用主成分分析,可以观察出引起样本变化的主要因子。
标准化处理 此处我们采用了The trimmed mean of M-values normalization method (TMM) 去校正文库之间的组成偏好。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 > dgeObj <- calcNormFactors(dgeObj) > dgeObj$samples group lib.size norm.factors MCL1.DG 1 23218026 1.2368993 MCL1.DH 1 21768136 1.2139485 MCL1.DI 1 24091588 1.1255640 MCL1.DJ 1 22656713 1.0698261 MCL1.DK 1 21522033 1.0359212 MCL1.DL 1 20008326 1.0872153 MCL1.LA 1 20384562 1.3684449 MCL1.LB 1 21698793 1.3653200 MCL1.LC 1 22235847 1.0047431 MCL1.LD 1 21982745 0.9232822 MCL1.LE 1 24719697 0.5291015 MCL1.LF 1 24652963 0.5354877
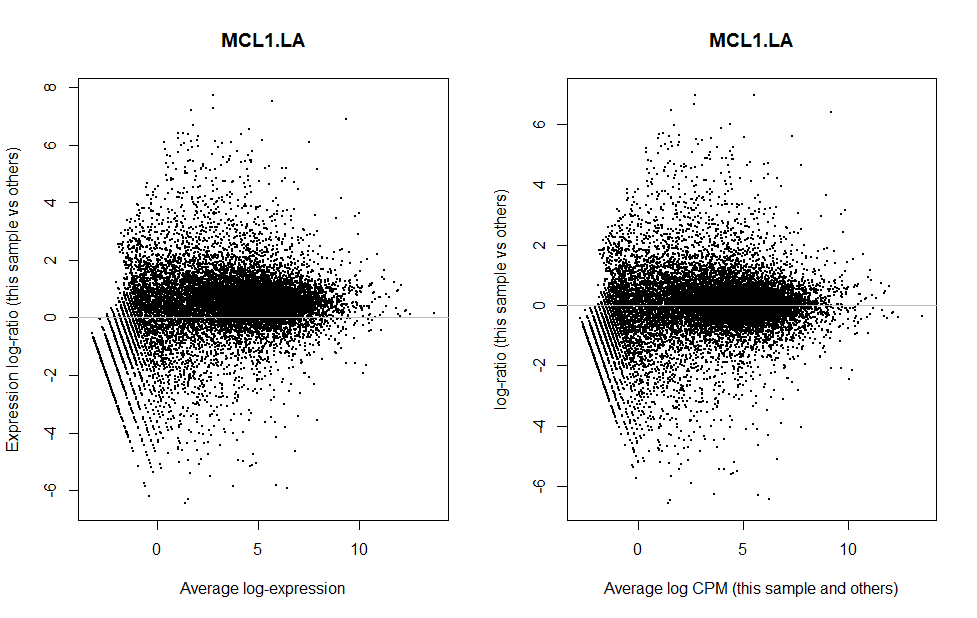
我们可以单独抽一个样本出来看看校正前后的数据分布情况
可以看出校正后,样本表达更加集中于0。
至此,数据的预处理也已经完结.
1 2 save(group,dgeObj,sampleinfo,file="~preprocessing.Rdata" )
Step 3: Differential Expression for RNA-seq本节将要介绍的是在R中进行RNA-seq数据基因表达差异分析的实战代码.
Ref: https://bioinformatics-core-shared-training.github.io/RNAseq-R/rna-seq-de.nb.html
在预处理生成了表达矩阵且标准化后,接下来我们要做的就是差异分析了。
本次流程需要的包
同时载入上次预处理完的数据preprocessing.Rdata
1 2 3 4 5 6 library(edgeR) library(limma) library(Glimma) library(gplots) library(org.Mm.eg.db) load("preprocessing.Rdata" )
在edgeR包的分析流程中,构建了表达矩阵后,便需要使用model.matrix函数创建保存有分组信息的矩阵design matrix 。该矩阵以$\color{red}{0/1}$的方式存储了分组信息。本次分析中的分组信息包括了:小鼠状态与细胞类型 ,现在我们先在两种因素并不存在相互作用 的假设下拟合线性模型。
1 2 3 4 5 > group <- as.character (group) > group [1 ] "basal.virgin" "basal.virgin" "basal.pregnant" "basal.pregnant" "basal.lactate" [6 ] "basal.lactate" "luminal.virgin" "luminal.virgin" "luminal.pregnant" "luminal.pregnant" [11 ] "luminal.lactate" "luminal.lactate"
group的分组信息包括了状态和细胞类型,因此我们使用strsplit函数,以‘.’为分隔把细胞类型信息和状态信息分别提取出来。
1 2 3 4 5 6 7 8 > type <- sapply(strsplit(group, "." , fixed=T ), function (x) x[1 ]) > status <- sapply(strsplit(group, "." , fixed=T ), function (x) x[2 ]) > type [1 ] "basal" "basal" "basal" "basal" "basal" "basal" "luminal" "luminal" "luminal" [10 ] "luminal" "luminal" "luminal" > status [1 ] "virgin" "virgin" "pregnant" "pregnant" "lactate" "lactate" "virgin" "virgin" [9 ] "pregnant" "pregnant" "lactate" "lactate"
成功提取后,构建分组信息矩阵
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 > design <- model.matrix(~ type + status) > design (Intercept) typeluminal statuspregnant statusvirgin 1 1 0 0 1 2 1 0 0 1 3 1 0 1 0 4 1 0 1 0 5 1 0 0 0 6 1 0 0 0 7 1 1 0 1 8 1 1 0 1 9 1 1 1 0 10 1 1 1 0 11 1 1 0 0 12 1 1 0 0 attr (,"assign" )[1 ] 0 1 2 2 attr (,"contrasts" )attr (,"contrasts" )$type[1 ] "contr.treatment" attr (,"contrasts" )$status[1 ] "contr.treatment"
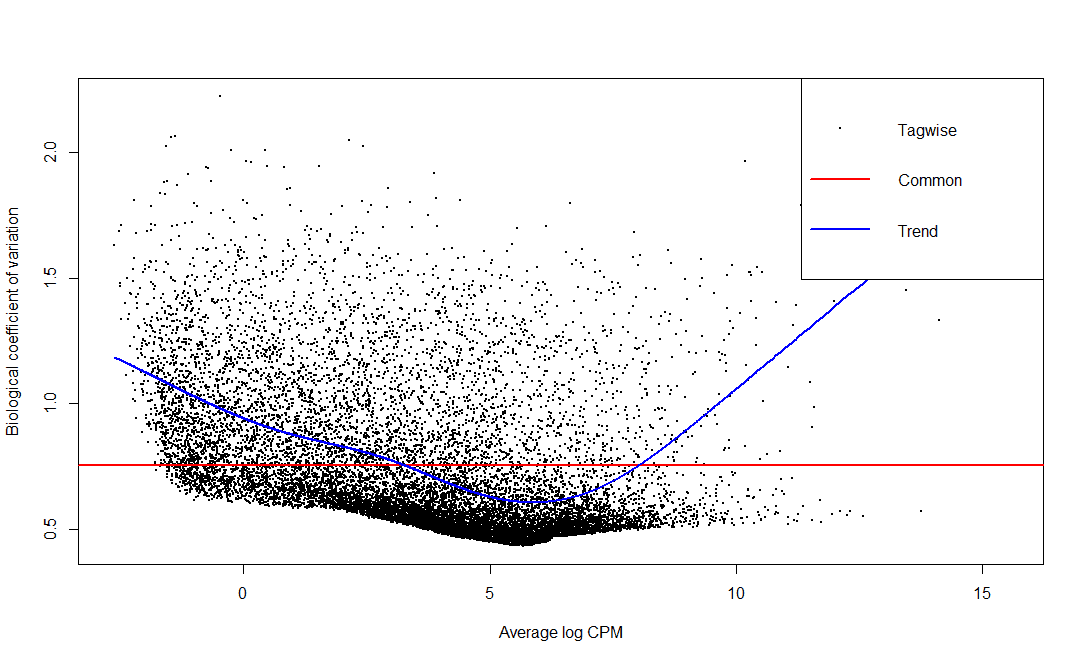
预测基因表达的离散度 这里根据每个基因在不同样本之间的表达量对基因表达的离散度(dispersion)进行预测。对基因表达离散度的预测将会帮助我们对各个样本的counts进行校正。
1 2 3 4 5 6 > dgeObj <- estimateCommonDisp(dgeObj) > dgeObj <- estimateGLMTrendedDisp(dgeObj) > dgeObj <- estimateTagwiseDisp(dgeObj) > plotBCV(dgeObj)
实际上以上三句代码可以用一个函数代替。在真实的分析当中也是使用estimateDisp()预测基因离散度,上面的拆分只是为了帮助我们理解离散度的预测是由哪几方面出发的。
1 dgeObj <- estimateDisp(dgeObj, design = design)
差异表达分析 预测离散度后,简单来说,我们将使用edgeR::glmFit()进行模型的拟合;使用edgeR::glmLRT()检验基因表达差异是否显著。
1 2 3 4 5 6 7 8 9 10 > fit <- glmFit(dgeObj, design) > head(coef(fit)) (Intercept) typeluminal statuspregnant statusvirgin 497097 -11.187922 -7.58804851 -0.7085514 -0.09305118 20671 -12.715063 -1.85287334 0.2269001 0.49554506 27395 -11.221391 0.56368066 -0.1415910 -0.29221577 18777 -10.146793 0.08280255 -0.1845489 -0.48795441 21399 -9.909825 -0.24195503 0.1753606 0.13494615 58175 -16.310131 3.09936215 1.1975518 0.84742701
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 > lrt.BvsL <- glmLRT(fit, coef=2 ) > topTags(lrt.BvsL) Coefficient: typeluminal logFC logCPM LR PValue FDR 110308 -8.940579 10.264297 24.89789 6.044844e-07 0.0004377961 50916 -8.636503 5.749781 24.80037 6.358512e-07 0.0004377961 12293 -8.362247 6.794788 24.68526 6.749827e-07 0.0004377961 56069 -8.419433 6.124377 24.41532 7.764861e-07 0.0004377961 24117 -9.290691 6.757163 24.32506 8.137331e-07 0.0004377961 12818 -8.216790 8.172247 24.24233 8.494462e-07 0.0004377961 22061 -8.034712 7.255370 24.16987 8.820135e-07 0.0004377961 12797 -9.001419 9.910795 24.12854 9.011487e-07 0.0004377961 50706 -7.697022 10.809629 24.06926 9.293193e-07 0.0004377961 237979 -8.167451 5.215921 24.03528 9.458678e-07 0.0004377961 de.res <- topTags(lrt.BvsL, n = Inf )
最后,保存差异分析的结果.
1 save(lrt.BvsL,dgeObj,group,file="DE.Rdata" )
Step 4: Annotation and Visualisation of RNA-seq results本节将介绍RNA-seq差异分析结果可视化的实战代码。
Ref: https://bioinformatics-core-shared-training.github.io/RNAseq-R/rna-seq-annotation-visualisation.nb.html
在差异分析后我们经常要可视化我们的分析结果,例如我们可以使用火山图将差异表达的基因画出来。
基因注释 在本次分析中,我们都是采用基因的ID来进行的,但如果想便于我们自己查阅的话,单纯的ID是比较麻烦的,我们还得一个个ID找它对应的基因名。因此,我们可以在差异分析结果的表格中添加上基因的symbol,genename等信息。
由于这次分析的物种是小鼠,我们采用以下的注释包:
1 2 3 4 > library(org.Mm.eg.db) > load("DE.Rdata" )
让我们查看一下该注释包存有的数据
1 2 3 4 5 > keytypes(org.Mm.eg.db) [1 ] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS" "ENTREZID" [7 ] "ENZYME" "EVIDENCE" "EVIDENCEALL" "GENENAME" "GO" "GOALL" [13 ] "IPI" "MGI" "ONTOLOGY" "ONTOLOGYALL" "PATH" "PFAM" [19 ] "PMID" "PROSITE" "REFSEQ" "SYMBOL" "UNIGENE" "UNIPROT"
在以上的数据中,我们需要的**”ENTREZID”** 、**”GENENAME”、 “SYMBOL”** 都在其中。
接下来,我们只要对应已有结果中的ENTREZID 提取相应的GENENAME 和SYMBOL 即可。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 > results <- as.data.frame(topTags(lrt.BvsL,n = Inf )) > ann <- select(org.Mm.eg.db,keys=rownames(results), + columns=c ("ENTREZID" ,"SYMBOL" ,"GENENAME" )) 'select()' returned 1 :1 mapping between keys and columns> head(ann) ENTREZID SYMBOL GENENAME 1 110308 Krt5 keratin 5 2 50916 Irx4 Iroquois homeobox 4 3 12293 Cacna2d1 calcium channel, voltage-dependent, alpha2/delta subunit 1 4 56069 Il17b interleukin 17 B5 24117 Wif1 Wnt inhibitory factor 1 6 12818 Col14a1 collagen, type XIV, alpha 1
利用cbind函数将新的注释信息与原表格合并之后即可
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 > results.annotated <- cbind(results, ann) > head(results.annotated) logFC logCPM LR PValue FDR ENTREZID SYMBOL 110308 -8.940579 10.264297 24.89789 6.044844e-07 0.0004377961 110308 Krt550916 -8.636503 5.749781 24.80037 6.358512e-07 0.0004377961 50916 Irx412293 -8.362247 6.794788 24.68526 6.749827e-07 0.0004377961 12293 Cacna2d156069 -8.419433 6.124377 24.41532 7.764861e-07 0.0004377961 56069 Il17b24117 -9.290691 6.757163 24.32506 8.137331e-07 0.0004377961 24117 Wif112818 -8.216790 8.172247 24.24233 8.494462e-07 0.0004377961 12818 Col14a1 GENENAME 110308 keratin 5 50916 Iroquois homeobox 4 12293 calcium channel, voltage-dependent, alpha2/delta subunit 1 56069 interleukin 17 B24117 Wnt inhibitory factor 1 12818 collagen, type XIV, alpha 1
此外,除了基因名的信息之外,还可以利用有基因组位点信息的包,如TxDb.Mmusculus.UCSC.mm10.knownGene(小鼠的)和GenomicRanges来注释基因座信息。
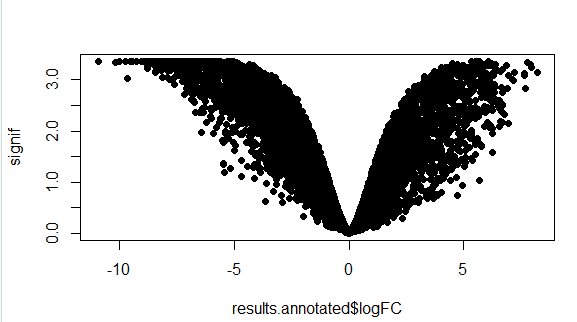
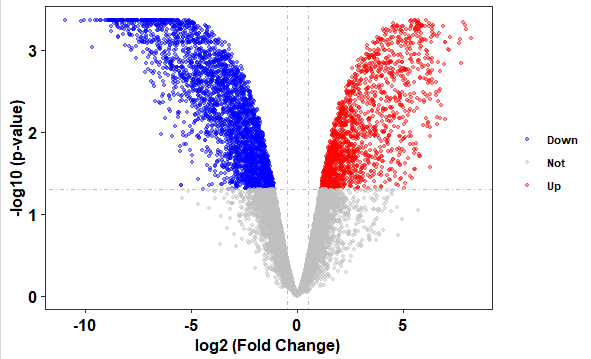
可视化 火山图是一种常用的差异表达分析结果可视化形式。该图横轴展示基因表达差异倍数,纵轴表示基因表达差异的显著性。
先看看最简单的图
1 2 > signif <- -log10(results.annotated$FDR) > plot(results.annotated$logFC,signif ,pch=16 )
当然,各位R学习者肯定不会止步于画了这个图就算了。让我们用ggplot画出更漂亮的图吧!
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 > library(ggplot2) > library(ggtheme) > results.annotated$threshold <- as.factor( + ifelse(results.annotated$FDR < 0.05 & abs (results.annotated$logFC) >=log2(2 ), + ifelse(results.annotated$logFC > log2(2 ) ,'Up' ,'Down' ),'Not' )) > plt <- ggplot(data=results.annotated, aes(x=logFC, y =-log10(FDR), colour=threshold,fill=threshold)) + + scale_color_manual(values=c ("blue" , "grey" ,"red" ))+ + geom_point(alpha=0.4 , size=1.2 ) + + theme_bw(base_size = 12 , base_family = "Times" ) + + geom_vline(xintercept=c (-0.5 ,0.5 ),lty=4 ,col="grey" ,lwd=0.6 )+ + geom_hline(yintercept = -log10(0.05 ),lty=4 ,col="grey" ,lwd=0.6 )+ + theme(legend.position="right" , + panel.grid=element_blank(), + legend.title = element_blank(), + legend.text= element_text(face="bold" , color="black" ,family = "Times" , size=8 ), + plot.title = element_text(hjust = 0.5 ), + axis.text.x = element_text(face="bold" , color="black" , size=12 ), + axis.text.y = element_text(face="bold" , color="black" , size=12 ), + axis.title.x = element_text(face="bold" , color="black" , size=12 ), + axis.title.y = element_text(face="bold" ,color="black" , size=12 )) + + labs( x="log2 (Fold Change)" ,y="-log10 (p-value)" ) > plt
除了火山图,还有各种对差异分析结果可视化的图就不在此展开了,有兴趣的朋友可以到原网站上继续了解。
Step 5: Gene-set testing本节将介绍基因富集分析的实战代码。
Ref: https://bioinformatics-core-shared-training.github.io/RNAseq-R/rna-seq-gene-set-testing.nb.html
在基因差异分析后,我们会得到一个包含了差异表达基因信息的表格。但有时过于冗长的 基因列表或是繁杂的 信息会阻碍我们从中提取有效的信息。因此,科学家们时常会采用基因富集分析的办法,去掌握差异表达基因 真正影响到的信号通路 或是代谢途径 等。
例如,Gene Ontology(GO) 或是KEGG 就是较为常用的基因富集分析,不仅可以帮助我们从冗长的数据中提取出有生物学意义的信息 ,还能有助于我们从系统的水平 观察细胞/机体的变化。
接下来,我们就以GOseq这个包向大家展示在R中处理的GO富集分析流程。当然我们只是简要地介绍一下,如果想要深入了解的朋友,可以查看Bioconductor上的帮助文档:GOseq
本次流程所需要的包和数据集
1 2 3 4 5 library(edgeR) library(goseq) load("DE.Rdata" )
GOseq analysis pipeline 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 results <- as.data.frame(topTags(lrt.BvsL, n = Inf )) genes <- results$FDR < 0.01 names (genes) <- rownames(results)pwf <- nullp(genes, "mm10" ,"knownGene" ) go.results <- goseq(pwf, "mm10" ,"knownGene" ) head(go.results,10 ) category over_represented_pvalue under_represented_pvalue numDEInCat numInCat 16121 GO:0071944 4.062458e-63 1 933 3543 2723 GO:0005886 2.686421e-62 1 908 3420 2540 GO:0005576 4.492776e-57 1 469 1457 11253 GO:0044421 1.026496e-50 1 391 1175 11283 GO:0044459 2.158946e-49 1 563 1893 2562 GO:0005615 2.018827e-47 1 336 975 7483 GO:0031224 2.789374e-41 1 833 3414 7485 GO:0031226 3.115084e-40 1 323 923 2724 GO:0005887 7.600430e-40 1 309 867 4426 GO:0009653 1.471331e-39 1 592 2186 term ontology 16121 cell periphery CC2723 plasma membrane CC2540 extracellular region CC11253 extracellular region part CC11283 plasma membrane part CC2562 extracellular space CC7483 intrinsic component of membrane CC7485 intrinsic component of plasma membrane CC2724 integral component of plasma membrane CC4426 anatomical structure morphogenesis BP
GO的分析也就到此为止了,至于后续的可视化的话就不先在此展开了。实际上还有许多R包也能在R中进行基因富集分析,例如Y叔的clusterProfiler,fgsea,camera等等。
关于RNA-seq分析流程到这就先告一段落了,文章中有任何错误都请各位小伙伴指出,共同学习!
完。