整理ChIP-seq / CUT & Tag 分析时用到的工具。本文只对使用的工具用法进行简单介绍。
当我们需要评估ChIP-seq类测序数据的相关性时,deeptools 是一个可行且方便的工具。它提供了一系列方便的命令对高通量测序数据进行分析。本文先集中介绍deeptools中计算ChIP-seq样本间相关性所用到的命令,其余的命令有机会再一一介绍。
multiBamSummary/multiBigwigSummary
multiBamSummary / multiBigwigSummary 两个命令可以分别计算 bam/bw 文件在基因组或特定区域测序覆盖度。
下面的命令将基因组以bin size (eg: 500bp) 为单位分割,计算bam文件在基因组每个bins上的read counts,并以.npz的格式输出,供后面作图使用。另外,使用参数--outRawCounts将read counts保存到文件readCounts.tab中
1 | multiBamSummary bins --bamfiles file1.bam file2.bam -o results.npz -bs <bin size> --outRawCounts readCounts.tab |
Example
使用ENCODE ChIP-Seq 数据作为示例
1 | $ deepTools2.0/bin/multiBigwigSummary bins \ |
或者对bw 文件进行计算
1 | multiBigwigSummary bins -b file1.bw file2.bw -o results.npz |
plotCorrelation
plotCorrelation 命令根据multiBamSummary / multiBigwigSummary输出的.npz文件计算样本之间的Pearson or Spearman 相关系数,并进行热图或散点图的绘制。
还是以上面ENCODE data为例
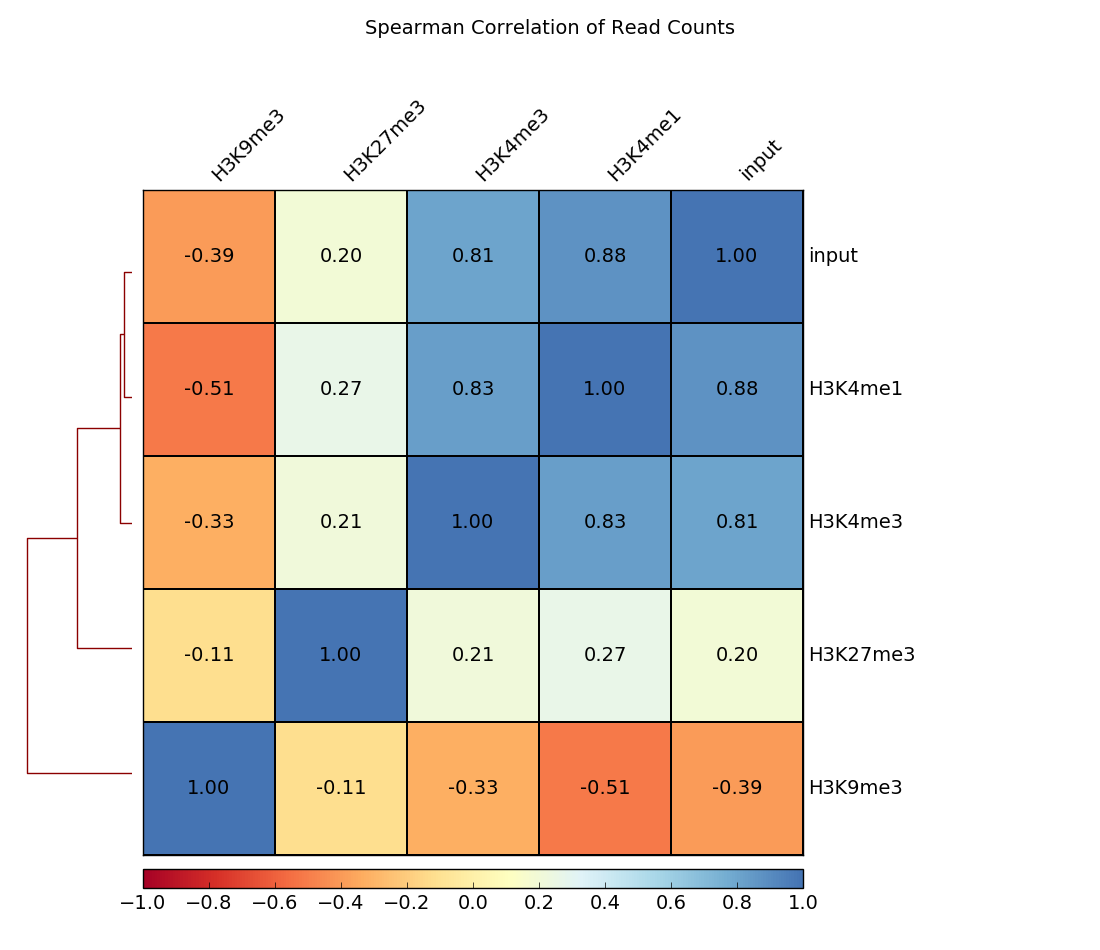
使用热图展示样本间相关性
1 | $ deepTools2.0/bin/plotCorrelation \ |
使用散点图展示样本间相关性,图内每个点代表一个bin,横纵坐标表示相应样本的bins内的counts数目
1 | $ deepTools2.0/bin/plotCorrelation \ |
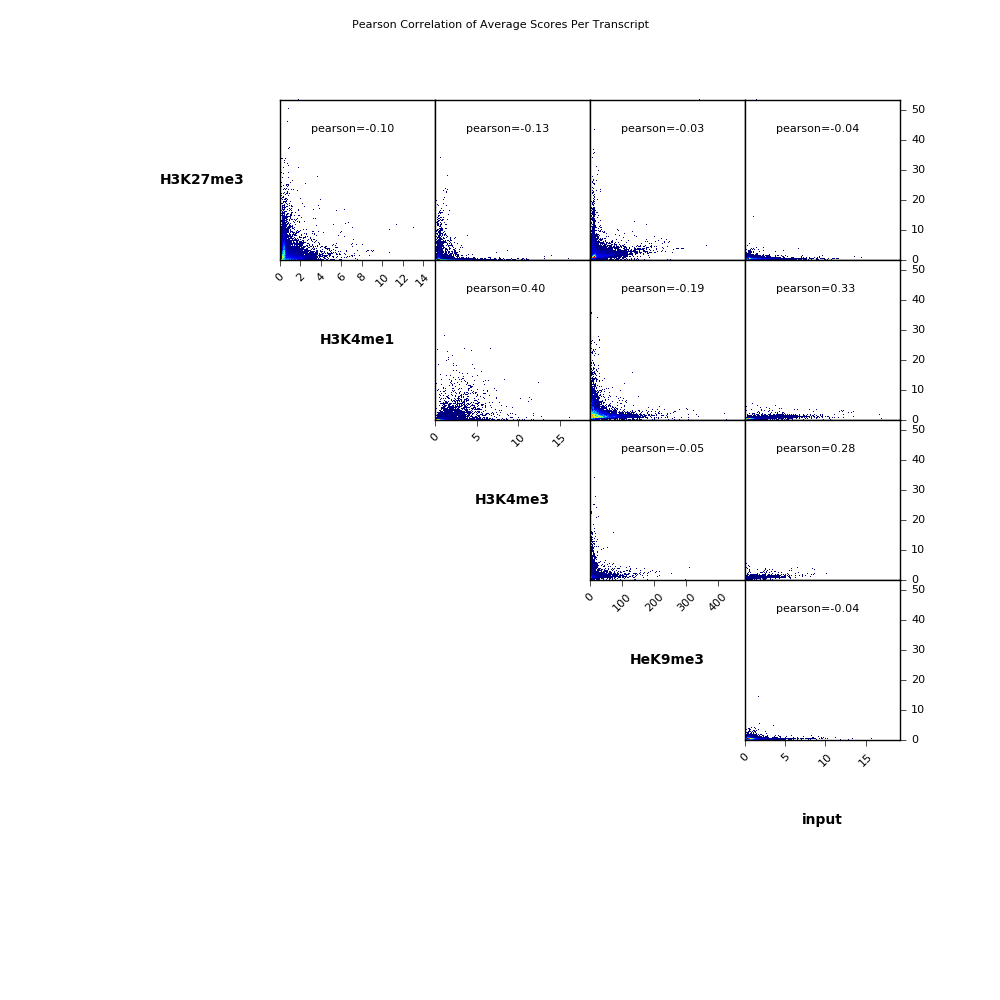
-in :指定输入的.npz文件 (由multiBamSummary / multiBigwigSummary输出)--corMethod:指定相关系数,Pearson or Spearman--skipZeros:跳过数据中含0的行计算相关系数--whatToPlot:绘制的图的种类,heatmap or scatterplot-o:图片输出的名称--outFileCorMatrix:输出相关系数矩阵
以上就是使用deeptools进行ChIP-seq样本间相关性计算的简单整理,关于计算相关系数之间的normalization和相关系数的选择在此不作展开,以后有机会再写一篇来谈吧。
Ref:
https://deeptools.readthedocs.io/en/develop/content/tools/multiBamSummary.html
https://deeptools.readthedocs.io/en/develop/content/tools/multiBigwigSummary.html
https://deeptools.readthedocs.io/en/develop/content/tools/plotCorrelation.html
完。