在高通量测序数据的分析中,仅仅靠raw read counts描述基因的表达量是远远不够的。受限于测序过程中的技术因素影响,read counts对于基因表达量的反映存在一定偏好(bias)。因此,Mortazavi等人提出了RPKM/FPKM的方法对read counts进行normalization以使基因表达量的比较可以在不同文库间进行。随后,Mortazavi等人更是提出了考虑转录本长度分布情况的TPM方法。本文将会简要说明为什么我们要对read counts进行normalization,以及RPKM,FPKM,TPM是什么,并通过一个简单地例子阐述为什么TPM才是被更多人认同的方法。
为什么我们要进行Normalization
我们之所以说测序得到的read counts并不是其mRNA丰度的忠实反映,是因为read counts的数量会受到多种因素的影响,例如:
测序深度:测序深度直接影响样本的read counts。例如,测序深度高的样本,同一个基因的read counts相对测序深度较低的样本而言也会较高,即使这个基因并没有发生差异表达。另外,某些低表达量的基因只有在较高的测序深度时才能检测到。一般而言,随着测序深度的增加,基因种类以及可变剪接体的数目也会增加。

在上图中,样本A中的基因X表达量是样本B的两倍,但这是由于测序深度引起的结果,而非真实存在的差异。
基因长度:由于高通量测序是将cDNA碎片化后再进行测序的,因此越长的基因产生的片段会越多,其read counts也更多。这就导致了较长的基因read counts比较短的基因要更多,从而混淆了不同长度基因间表达量的比较,所以对基因长度的校正也是十分有必要的。

在这个图里,基因X和Y的真实表达量是一致的,但是基因X的read counts会比基因Y要多,这是由于基因X的基因长度较长所致的。
除了上述两个主要因素外,还会有其他因素对read counts的检测有所影响,例如转录组的组成,GC含量,random hexamers引起的测序偏好等等。由于上述因素的存在,导致在不同样本间使用read counts 进行比较是不太现实的,人们便提出了许多对read counts进行normalization的方法。本文在此简单地介绍使用最为广泛却最受质疑的RPKM/FPKM,以及受到更多人认可的TPM。
RPKM/FPKM
RPKM: Reads per kilo base per million mapped reads (single-end sequencing)
FPKM: Fragments per kilo base per million mapped reads (paired-end sequencing)
RPKM与FPKM实际上一样的单位,只不过RPKM是在单端测序文库中使用,而FPKM是双端测序所用的
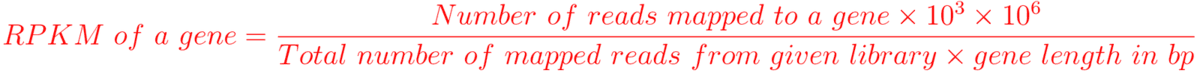
从RPKM的公式中我们可以看到,RPKM对基因长度(gene length)以及测序深度(mapped reads from library)都进行了校正。而FPKM只是RPKM的双端测序(pair-end)版本。
其R语言实现如下:
1 | RPKM <- function(x, gene.length){ |
其中x为一个向量存储了每个基因的counts,而gene.length是相应于x中每个基因的长度的一个向量.
TPM
TPM: Transcript per million
TPM的计算公式如下所示:

同RPKM一样,TPM对基因的长度进行了校正,计算比对到基因上的reads/基因长度得到长度校正的表达量 reads per kilobase (RPK)。再以文库中RPK之和作为Scale Factor求出TPM。
其R语言实现如下:
1 | TPM <- function(x, gene.length){ |
其中x为一个向量存储了每个基因的counts,而gene.length是相应于x中每个基因的长度的一个向量.
相比于RPKM使用文库大小(read counts之和)来作为文库校正因子,TPM使用RPK之和作为文库校正因子的好处是考虑了不同样本间的基因长度的分布。因为RPK是一个对基因长度进行校正后的表达量单位,所以RPK之和也不会再带入基因长度的bias。因此,如果需要比较的样本之间转录本分布不一致时(例如不同物种RNA-seq的比较),使用TPM是一个较佳的normalization方案。
例子:RPKM/FPKM vs TPM
接下来,我们使用一个例子展示RPKM/FPKM和TPM方法校正的区别。
以下有ABCD四个基因,并同时进行了三次重复的测序(rep1,rep2,rep3)。首先,可以看出由于基因长度的关系,B基因的read counts都是较其余的基因较高的(排除D而言)。其次,可以看出rep3的测序深度较高,得到的read counts也较多,同时还检测到其余两个重复中没有检测到的D基因(低丰度基因)。不管是测序深度还是基因长度的严重地干扰了我们对不同样本的基因表达量比较。因此,我们采用RPKM和TPM的方法进行Normalization后,再来比较其中的差异。
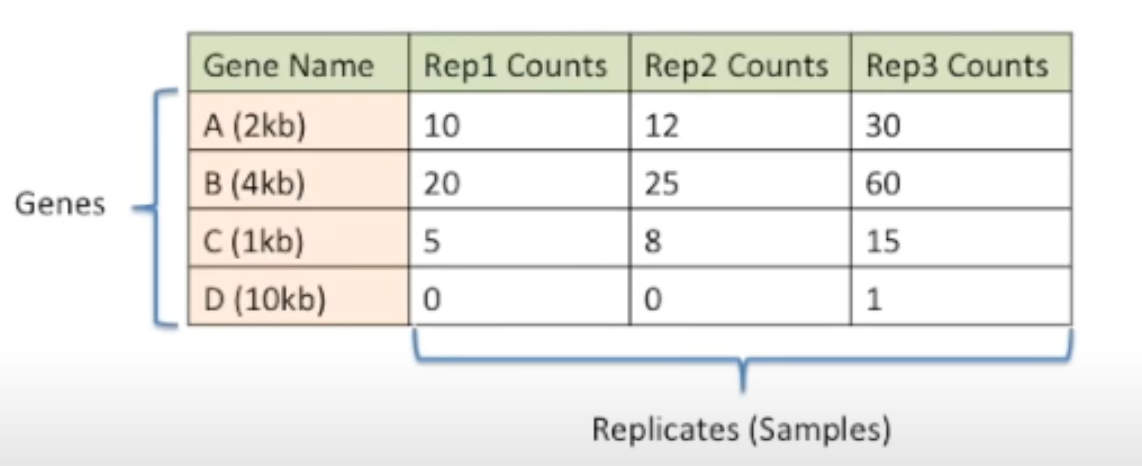
为了方便起见,以下的函数省略了kilo base,million base的转换
1 | RPKM <- function(x, gene.length){ |
通过以上的例子,我们可以发现RPKM和TPM最重要且直接的区别就是,TPM校正后的表达量之和在不同样本中是相等的,而在RPKM中并不是。
由于RPKM在各样本之和不一致,导致我们不能直接比较同一基因在不同样本间的RPKM。例如,对于基因A而言,RPKM校正后,其在rep1和rep2中的占比分别为142.8571/428.5714和133.3333/450。相反,由于TPM在各样本之和是一致的,我们可以直接比较不同样本的TPM来比较该基因表达量在样本中的占比。
简而言之,RPKM和TPM这类方法就是为了使不同样本间的总体表达量趋于一致,让不同样本间的基因表达量有可比较性,而TPM能够更好地校正样本间的差异。
- 常用的Normalization 方法总结
| Normalization method | 描述 | 考虑因素 | 使用场景 |
|---|---|---|---|
| CPM (counts per million) | 使用read counts的总和校正counts | 测序深度 | 同一样本组重复样本之间的基因counts比较;不适用于样品内的比较或差异分析 |
| TPM (transcripts per kilobase million) | 每百万mapped reads中每kb转录本上的reads数 | 测序深度和基因长度 | 样本内或同一样本组样本之间的基因counts比较; 不适用于差异分析 |
| RPKM/FPKM (reads/fragments per kilobase of exon per million reads/fragments mapped) | 如TPM | 测序深度和基因长度 | 样本内基因间的counts比较; 不适用于样本间比较和差异分析 |
| DESeq2’s median of ratios [1] | counts除以样本特异的校正因子,该因子由基因计数相对于每个基因的几何平均值的中位数比率确定 | 测序深度及RNA组成 | 样本之间的基因counts比较以及差异分析; 不适用于样本内比较 |
| EdgeR’s trimmed mean of M values (TMM) [2] | 使用样本之间的加权截尾的对数表达量比值的均值进行TMM校正 | 测序深度,RNA组成以及基因长度 | 样品之间和样品内部的基因counts比较,适用于差异分析 |
这里要补充的是在进行差异表达分析时,我们实际上并不会用到RPKM/FPKM, TPM,而是使用raw counts给到差异分析的工具。这是由于RPKM/FPKM和TPM都没有考虑到样本间文库组成的差异,因而不适合用于样本间差异分析。例如,直接比较小鼠肝脏与肾脏之间基因表达的TPM是不太合理的。
Ref:
- https://www.rna-seqblog.com/rpkm-fpkm-and-tpm-clearly-explained/
- https://www.biostars.org/p/273537/
- https://www.jianshu.com/p/1940c5954c81
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008 Jul 1;5(7):621-8.
- https://hbctraining.github.io/DGE_workshop/lessons/02_DGE_count_normalization.html
完。