在做基因差异表达分析时,经常会用DESeq2这个包,但一直没有深究其分析的统计流程。因此,在这里记录一下DESeq2校正基因表达的方法 – 比率中值法 (Median of ratios)。
开篇明义,比率中值法考虑的因素是测序深度和文库的RNA组成。
以下我们使用airway数据具体展示比率中值法的校正过程
1 | library(airway) |
比率中值法具体实现如下:
Step 1: 创建假参考样本 (每个基因的几何平均值)
首先,我们为每一个基因创建一个假参考样本(pseudo-reference sample)表达量,这代表理想的基因表达量。该值通过计算每个基因的几何平均值得到
几何平均值:n个数的乘积开n次根
${\displaystyle \left(\prod {i=1}^{n}x{i}\right)^{\frac {1}{n}}={\sqrt[{n}]{x_{1}x_{2}\cdots x_{n}}}}$
1 | gm <- apply(df1, 1, function(x) exp(mean(log(x)))) |
| SRR1039508 | SRR1039509 | SRR1039512 | SRR1039513 | pseudo | |
|---|---|---|---|---|---|
| ENSG00000260166 | 0 | 0 | 0 | 0 | 0.000 |
| ENSG00000266931 | 0 | 0 | 0 | 0 | 0.000 |
| ENSG00000104774 | 1939 | 1906 | 2294 | 1205 | 1787.806 |
| ENSG00000267583 | 0 | 1 | 0 | 0 | 0.000 |
| ENSG00000227581 | 1 | 0 | 0 | 0 | 0.000 |
| ENSG00000227317 | 0 | 0 | 0 | 0 | 0.000 |
Step 2: 计算每一个样本相对于参考样本的比值
接着,对于样本的每一个基因,我们计算一个 sample/reference 的比值。
1 | # step 2: calculates ratio of each sample to the reference |
以第一行基因的ratio为例,写出具体计算过程:
| Row.names | SRR1039508 | SRR1039509 | SRR1039512 | SRR1039513 | pseudo | R_SRR1039508 | R_SRR1039509 | R_SRR1039512 | R_SRR1039513 |
|---|---|---|---|---|---|---|---|---|---|
| ENSG00000000460 | 60 | 55 | 40 | 35 | 46.36182 | 60/46.36182=1.2941683 | 55/46.36182=1.1863209 | 40/46.36182=0.8627789 | 35/46.36182=0.7549315 |
| ENSG00000000971 | 3251 | 3679 | 6177 | 4252 | 4209.97390 | 0.7722138 | 0.8738772 | 1.4672300 | 1.0099825 |
| ENSG00000002919 | 234 | 246 | 298 | 211 | 245.28016 | 0.9540111 | 1.0029348 | 1.2149372 | 0.8602408 |
| ENSG00000003436 | 4373 | 4183 | 5794 | 3230 | 4301.42236 | 1.0166405 | 0.9724690 | 1.3469963 | 0.7509144 |
| ENSG00000004660 | 95 | 55 | 59 | 35 | 57.31279 | 1.6575705 | 0.9596461 | 1.0294385 | 0.6106839 |
| ENSG00000004766 | 492 | 413 | 546 | 337 | 439.72831 | 1.1188727 | 0.9392163 | 1.2416758 | 0.7663823 |
由于大部分基因不会发生差异表达,所以样本内的ratios应是相当的。
Step 3: 计算每个样本的校正因子
样本内(column-wise)所有比率的中值即为校正因子(normalization factor), 也就是DESeq2 的size factor
1 | # step 3: size factor |
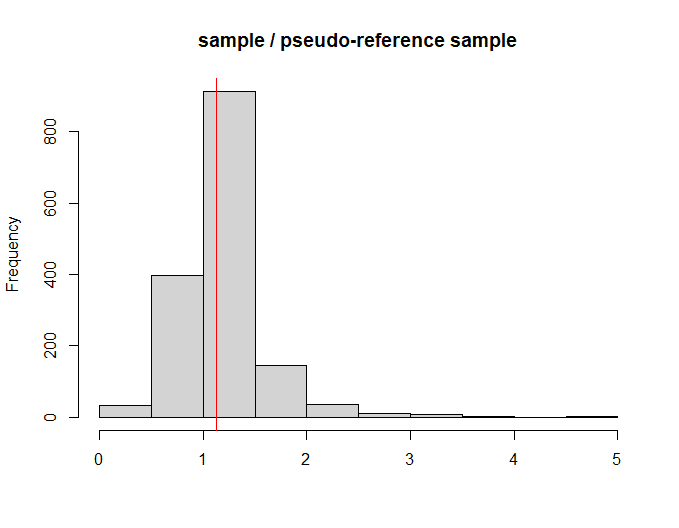
这里展示一个样本基因比率分布图,红线即为比率的中值
2
3
abline(v=median(df2$R_SRR1039508), col='red')
Step 4: 计算normalized coutns
1 | df1_norm <- t(t(df1[,-5])/sfs) |
| SRR1039508 | SRR1039509 | SRR1039512 | SRR1039513 | |
|---|---|---|---|---|
| ENSG00000260166 | 0.0000000 | 0.000000 | 0.000 | 0.000 |
| ENSG00000266931 | 0.0000000 | 0.000000 | 0.000 | 0.000 |
| ENSG00000104774 | 1724.8830895 | 1926.719981 | 1800.571 | 1671.653 |
| ENSG00000267583 | 0.0000000 | 1.010871 | 0.000 | 0.000 |
| ENSG00000227581 | 0.8895735 | 0.000000 | 0.000 | 0.000 |
| ENSG00000227317 | 0.0000000 | 0.000000 | 0.000 | 0.000 |
我们可以与DESeq2的sizefactor对比一下,看看我们算的是否正确
1 | # using deseq2 |
以上就是DESeq2计算size factors,以及校正counts的方法。事实上DESeq2在差异分析时并不会真正地用normalized counts进行分析,而是使用raw counts,并将size factor作为其广义线性回归模型(generalized linear model)的一个因子进行考虑。
完。
ref:
https://hbctraining.github.io/DGE_workshop/lessons/02_DGE_count_normalization.html