Neighbor Joining是一种bottom-up的聚类方法,常被用于**系统发育树(phylogenetic tree)*的构建当中。 Naruya Saitou 和 Masatoshi Nei在1987年将NJ法发表在Molecular Biology and Evolution*中,至今已有超5万的引入量,实在是生物信息学中超重量级的文章。

P.S. 由于本人对图论一知半解,所以后面写的时候可能各种术语穿插使用,望见谅。
术语阐明
taxa = node = 节点
edge = 边
算法原理
NJ法要求输入的数据必须是**待聚类数据(taxa)**之间的距离信息,例如对多个物种进行NJ法建树的话,输入的就是物种之间的进化距离。
NJ法是一种bottom-up的聚类,故首先要计算出进化距离最近的两个物种,将其聚为一类,再计算出距离该新类最近的一个物种再次聚为一个类,如此迭代,遍历所有输入的物种,构建系统发育树。
下面以对多个物种进行NJ法建树来简述其原理:
Q-criterion
对于n > 3的物种集合X而言,基于现有的距离矩阵 d(x, y)可以计算得到Q矩阵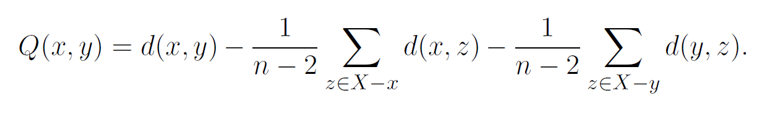
可将**Q(x,y)**视为计算一种矫正后的d(x,y)
选定x,y使得上述Q值最小,即为当前最靠近的两个taxa,而该法即为Q-criterion
构建新节点
找出了距离最近的(x,y)后,构建一个新的节点 Vx,y 取缔原有的两个节点(x,y),接下来可计算x,y点到新节点的距离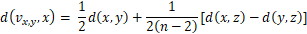
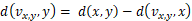
以及$V_{x,y}$ 到其余各点的距离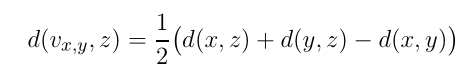
z: 在集合X中除x,y之外的各点
重新进行Q-criterion
上述两步结束后,就对集合X中最近的两点聚类完毕,继续迭代即可对集合中所有的点进行聚类,并构建发育树。
R应用的例子
这里用4个物种同源基因的进化距离矩阵进行NJ法建树,当然更一般来说我们会利用多重序列比对(Multiple Sequence Alignment,MSA),再根据MSA的结果得到进化距离矩阵构建系统发育树
现有4个物种的同源基因ABCD,它们的进化距离矩阵如下:
| | A | B | C | D |
| —- | —- | —- | —- | —- |
| A | 0 | | | |
| B | 5 | 0 | | |
| C | 12 | 11 | 0 | |
| D | 10 | 9 | 8 | 0 |
现将距离矩阵导入R中,使用R包ape的nj()函数建树
1 | library(ape) |
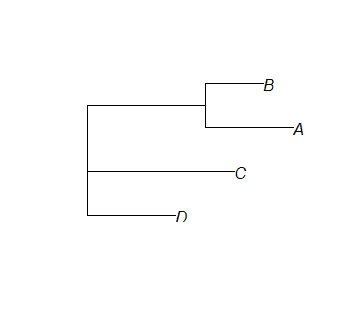
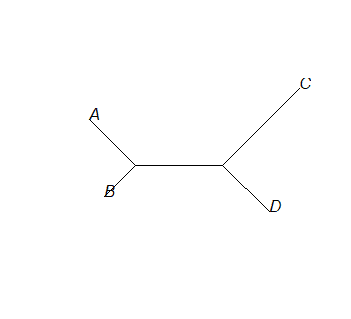
使用参数type可以控制输出有根树或无根树
1 | plot(tr, type = "unrooted", rotate.tree = 90) |
节点的距离,边的长度等信息,亦包含在nj()输出的结果中
1 | str(tr) |
利用R包ggtree可以将edge length 标在树上
1 | library(ggtree) |
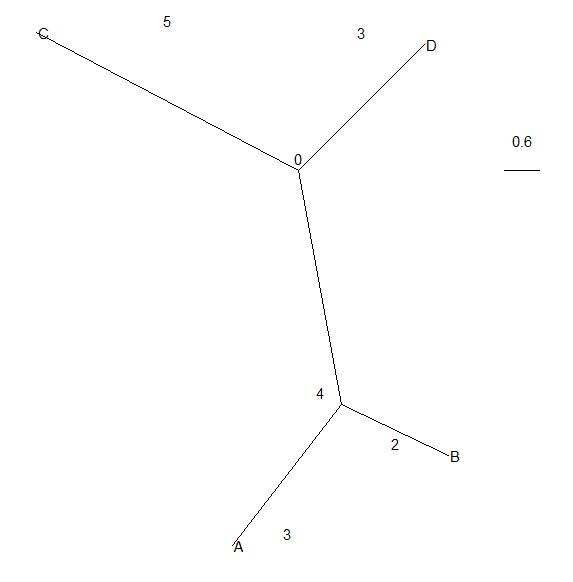
一点补充:
对同源基因/蛋白进行建树时,物种间的进化距离矩阵一般通过核酸/蛋白质序列的多序列比对(Multiple Sequence Algnment, MSA)结果而来。但如何从MSA的序列比对得分矩阵得到进化距离矩阵暂时还不清楚,以后有待补充。
Ref: en.wikipedia.org/wiki/Neighbor_joining
完。