整理ChIP-seq / CUT & Tag 分析时用到的工具。本文只对使用的工具用法进行简单介绍。
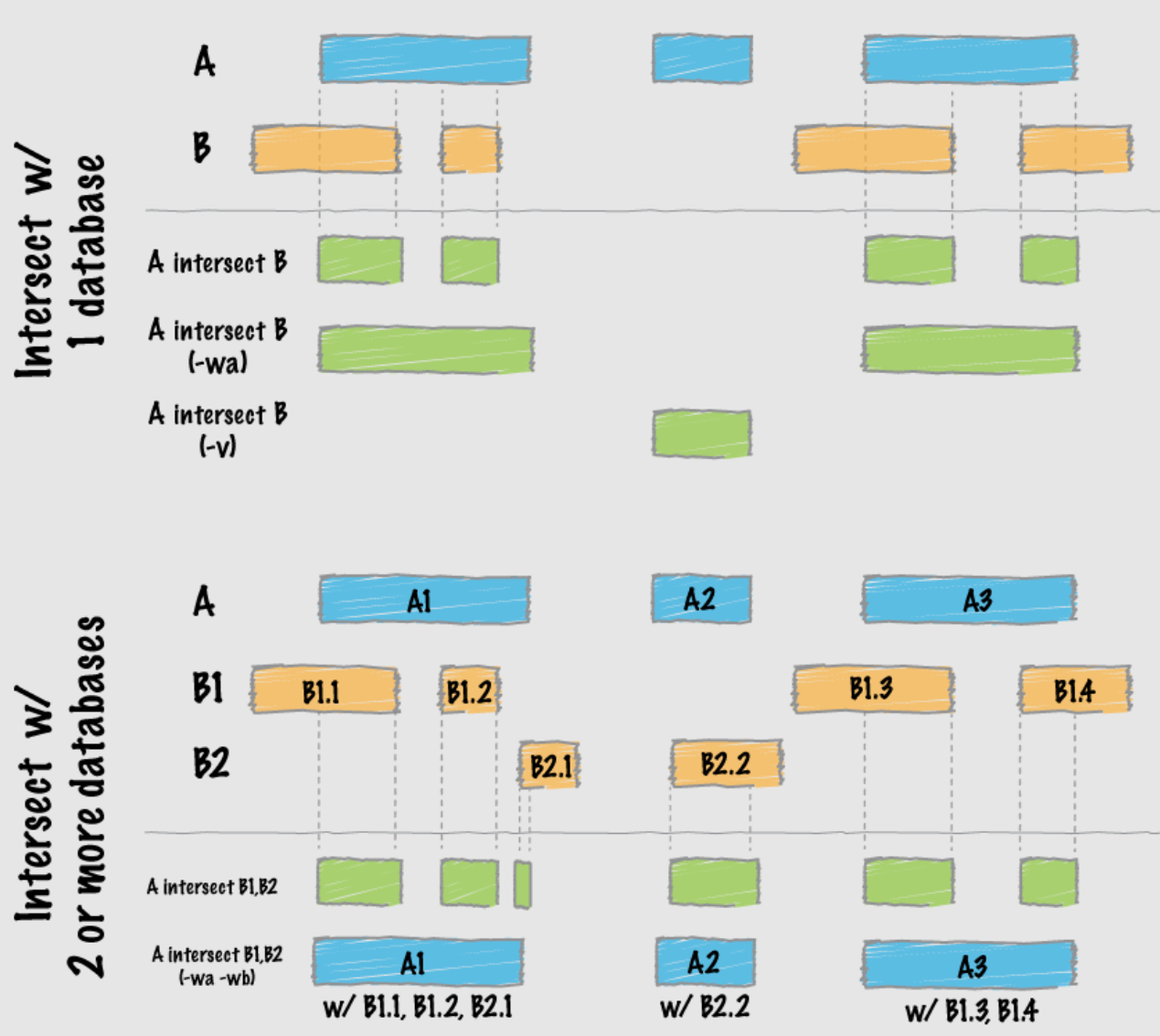
当我们想判断两个基因组区域,例如两个peaks是否重叠时,可以使用bedtools intersect解决这种问题。
以下使用bedtools官方提供的例子进行展示
一比一
一般情况,使用bedtools intersect寻找两个peaks文件的交集可以如下操作
1
2
3
4
5
6
7
8
9
| $ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed
chr1 15 20
|
-a:查询文件,可以是BAM/BED/GFF/VCF格式,A中的每一行都会与B进行比较以找出重叠的部分
-b:参考文件,依照-b输入的文件在A里面找重叠的部分,可以是BAM/BED/GFF/VCF格式。-b可以输入多个文件,用空格分开即可。现在可以用通配符(*)识别多个输入文件。
一比多
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
| $ cat query.bed
chr1 1 20
chr1 40 45
chr1 70 90
chr1 105 120
chr2 1 20
chr2 40 45
chr2 70 90
chr2 105 120
chr3 1 20
chr3 40 45
chr3 70 90
chr3 105 120
chr3 150 200
chr4 10 20
$ cat d1.bed
chr1 5 25
chr1 65 75
chr1 95 100
chr2 5 25
chr2 65 75
chr2 95 100
chr3 5 25
chr3 65 75
chr3 95 100
$ cat d2.bed
chr1 40 50
chr1 110 125
chr2 40 50
chr2 110 125
chr3 40 50
chr3 110 125
$ cat d3.bed
chr1 85 115
chr2 85 115
chr3 85 115
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-filenames
chr1 1 20 d1.bed chr1 5 25
chr1 40 45 d2.bed chr1 40 50
chr1 70 90 d1.bed chr1 65 75
chr1 70 90 d3.bed chr1 85 115
chr1 105 120 d2.bed chr1 110 125
chr1 105 120 d3.bed chr1 85 115
chr2 1 20 d1.bed chr2 5 25
chr2 40 45 d2.bed chr2 40 50
chr2 70 90 d1.bed chr2 65 75
chr2 70 90 d3.bed chr2 85 115
chr2 105 120 d2.bed chr2 110 125
chr2 105 120 d3.bed chr2 85 115
chr3 1 20 d1.bed chr3 5 25
chr3 40 45 d2.bed chr3 40 50
chr3 70 90 d1.bed chr3 65 75
chr3 70 90 d3.bed chr3 85 115
chr3 105 120 d2.bed chr3 110 125
chr3 105 120 d3.bed chr3 85 115
|
-wa:在交集中输出A的完整区间,而不仅是重叠的部分
-wb:在交集中输出B的完整区间,而不仅是重叠的部分
-sorted:该参数表明输入的文件是预先按照染色体和染色体坐标排序过,可以提高bedtools运行的效率。
-filenames:标明重叠区域是源于和哪个B文件发生重叠。
输出A unique的区域
1
2
3
4
5
6
7
| $ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-v
chr3 150 200
chr4 10 20
|
-v:输出A unique的区域
定义重叠阈值
这里定义两个区域100%一样才算重叠
1
2
3
4
5
6
7
8
9
| $ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-names d1 d2 d3
-f 1.0
chr1 40 45 d2 chr1 40 50
chr2 40 45 d2 chr2 40 50
chr3 40 45 d2 chr3 40 50
|
-f:指定两个区域最小重叠多少bp才算与A发生重叠,默认1bp(1E9)
-F:指定两个区域最小重叠多少bp才算与B发生重叠,默认1bp(1E9)
输出重叠区域的长度
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
| $ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -wo
chr1 10 20 chr1 15 20 5
chr1 10 20 chr1 18 25 2
$ bedtools intersect -a A.bed -b B.bed -wao
chr1 10 20 chr1 15 20 5
chr1 10 20 chr1 18 25 2
chr1 30 40 . -1 -1 0
|
-wo:输出A、B重叠区域重叠的bp
-wao:输出A、B重叠区域重叠的bp,假如B中不存在A的这个区域则输出0。
合并输出结果中重叠的区域
有时候A与B的交集区域之间可能也有重叠,这一点可以通过-u参数解决。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
| $ cat A.bed
chr1 10 20
$ cat B.bed
chr1 15 20
chr1 17 22
$ bedtools intersect -a A.bed -b B.bed
chr1 15 20
chr1 17 20
$ bedtools intersect -a A.bed -b B.bed -u
chr1 10 20
|
输出A与B overlap 的次数
如果我们只是想知道A文件中的区域与B文件的区域发生了多少次重叠,可以使用-c参数,或者使用-C参数分别输出不同来源的overlaps
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
| cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ cat C.bed
chr1 16 21
chr1 19 26
$ bedtools intersect -a A.bed -b B.bed C.bed -c
chr1 10 20 4
chr1 30 40 0
$ bedtools intersect -a A.bed -b B.bed C.bed -C -filenames
chr1 10 20 B.bed 2
chr1 10 20 C.bed 2
chr1 30 40 B.bed 0
chr1 30 40 C.bed 0
|
考虑正负链
如果在找交集的过程中需要考虑链特异性的时候,可以使用-s参数;
或者使用-S参数,强制不考虑正负链,而只考虑基因组区域是否重叠
1
2
3
4
5
6
7
8
9
10
11
12
| $ cat A.bed
chr1 100 200 a1 100 +
$ cat B.bed
chr1 130 201 b1 100 -
chr1 132 203 b2 100 +
$ bedtools intersect -a A.bed -b B.bed -wa -wb -s
chr1 100 200 a1 100 + chr1 132 203 b2 100 +
$ bedtools intersect -a A.bed -b B.bed -wa -wb -S
chr1 100 200 a1 100 + chr1 130 201 b1 100 -
|
对bedtools intersect的介绍到此结束,bedtools的其他命令有机会再做介绍。
Ref:
bedtools intersect docs: https://bedtools.readthedocs.io/en/latest/content/tools/intersect.html
完。