经典的转录组差异分析通常会使用到三个工具limma/voom, edgeR和DESeq2, 今天我们同样使用一个小规模的转录组测序数据来演示edgeR的简单流程。
由于,edgeR和DESeq2都是使用基于负二项分布的广义线性回归模型(GLM)来对RNA-seq数据进行拟合和差异分析,所以我们都用同一个数据来分析。
文中使用的数据来自Standford 大学的一个拟南芥的small RNA-seq数据(https://bios221.stanford.edu/data/mobData.RData)
该数据包含6个样本:SL236,SL260,SL237,SL238,SL239,SL240, 并分成了三组,分别是:
MM=”triple mutatnt shoot grafted onto triple mutant root”
WM=”wild-type shoot grafted onto triple mutant root”
WW=”wild-type shoot grafted onto wild-type root”
简而言之,
WW组可以认为是实验的对照组,而MM和WM则是两个处理组。

P.S. 本文的分析是基于有生物学重复的单因子差异分析,关于无生物学重复或者多因子的情况,以后有机会再做展开。
对于edgeR的分析流程而言,我们需要输入的数据包括:
- 表达矩阵(
counts) - 分组信息(
group) - 拟合信息(
design):指明如何根据样本的分组进行建模
下面就以mobData 中的数据为例简单介绍edgeR 的分析流程
载入数据及生成DGEList
由于mobData 中的行名没有提供基因的ID,我们也不是为了探究生物学问题,就以mobData 的行数作为其ID
1 | library(edgeR) |
edgeR将数据存储在列表形式的DGEList对象中,需要指定的参数包括counts 和group . DGEList 将表达矩阵存储在$counts中,将样本的信息,例如分组情况和文库大小等存储在$samples中。
预处理
在进行差异分析之前,需要对样本数据的表达矩阵进行预处理,包括:
- 去除低表达量基因
- 探索样本分组信息 – 有助于挖掘潜在的差异样本
这里我们根据CPM normalization后的基因表达量作为过滤低表达基因的指标
1 | # cpm normalization |
edgeR默认使用 trimmed mean of M-values (TMM) 计算文库的scale factor进行normalization,以最大程度地缩小样本间基因表达量的log-fold change。这是因为TMM法认为样本间大部分的基因都没有发生差异表达,而那些真正差异表达的基因并不会受到normalization的严重影响。如此一来,便将那些由于测序引起的差异表达基因的表达量给校正了,消除了一部分的假阳性。
1 | degs.norm <- calcNormFactors(degs.keep, method = 'TMM') |
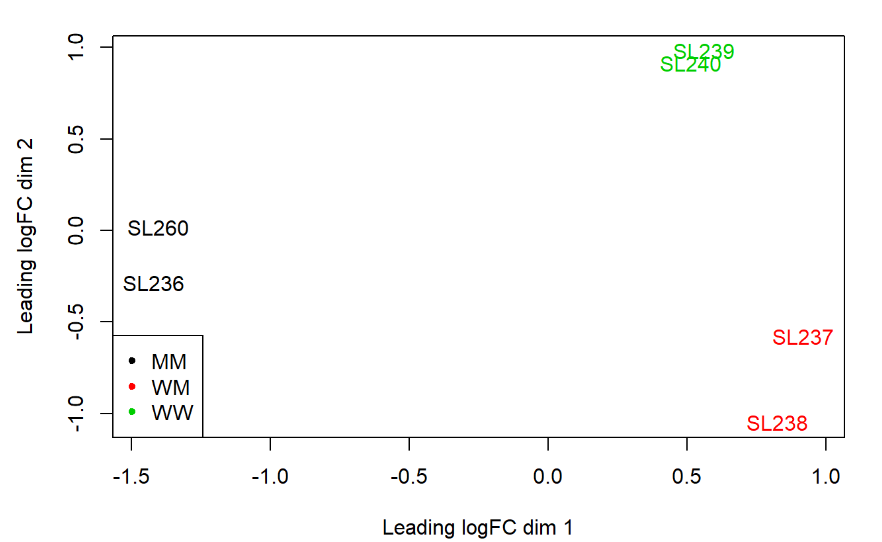
这里使用plotMDS 查看样本的分组情况(通过logFC),各组都分得很开。plotMDS在多因子的情况下可以更好地观察各个样本组是否有良好的分组。
关于Normalization
在差异分析中,我们常常更关注的是相对表达量的变化,例如处理组的A基因表达量相对于对照组的而言是上调还是下调了。而基因表达量的定量准确性则在差异分析中不太重要,因此,在进行差异分析时,像RPKM/FPKM这种对转录本长度进行normalization方法是并不常用,也是没有必要的。
在常规的RNA-seq中,影响基因表达量更大的技术因素往往是测序深度以及有效文库大小(effective libraries size)。这也是一般的差异分析软件会进行normalize的部分。
差异分析
首先,我们构建出design矩阵,指明差异分析所要比较的关系
1 | designMat <- model.matrix(~0+mobGroups);designMat |
然后,进行dispersion的估计
1 | degs.norm <- estimateGLMCommonDisp(degs.norm,design=designMat) |
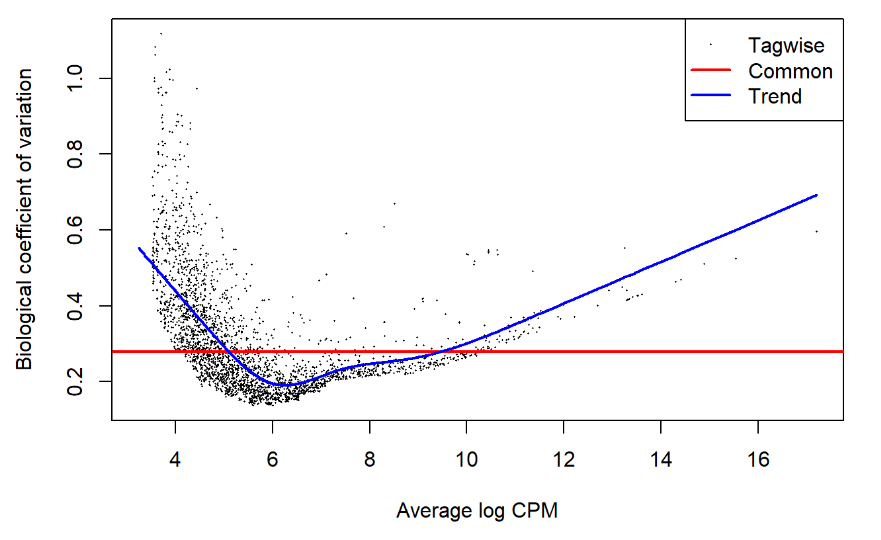
plotBCV 反映不同表达量的基因与模型拟合的情况,如果模型拟合得好则”Tagwise”点的分布会拟合到“Trend”这条曲线上,如上图所展示的情况。但也可以看到低表达量的基因有点离散。
关于dispersion estimation
一般而言,样本间的变异系数(coefficient of variance,CV)是由两部分组成的,一是技术差异(Technical CV),另一个是生物学差异(Biological coefficient of variance,BCV)。前者是会随着测序通量的提升而消失的,而后者则是样本间真实存在的差异。所以,对于一个基因g而言,它的BCV在样本间足够大的话,就可以认为基因g是一个差异表达基因。而
edgeR正是通过估计dispersion来估计BCV,进而拟合出线性回归模型的参数。
estimateGLMCommonDisp(x,design):为所有基因都计算同样的dispersion
estimateGLMTrendedDisp(x,design):根据不同基因的均值–方差之间的关系来计算dispersion,并拟合一个trended model
estimateGLMTagwiseDisp(x,design):计算每个基因的dispersion,并利用经验性贝叶斯方法(empirical bayes)压缩至Trend Didspersion。个人认为这一项相当于GLM中每个基因的beta值
最后,根据design进行拟合,并利用likelihood ratio test(LRT)进行统计检验
1 | fit <- glmFit(degs.norm, designMat) |
下面以MM组和 WW组的比较为例
topTags 提取出差异分析的数据,其中的contrast参数指定比较的组,1为“分子”,-1为“分母”,0即不考虑的变量;
decideTestsDGE 可以根据条件筛选差异基因,对每个基因分别赋予-1,0,1三个数值,分别代表下调,不显著和上调;
plotSmear 画一个简单的表达量与fold change的关系图。
1 | degs.res.1vs3 <- topTags(lrt.1vs3, n = Inf, adjust.method = 'BH', sort.by = 'PValue') |
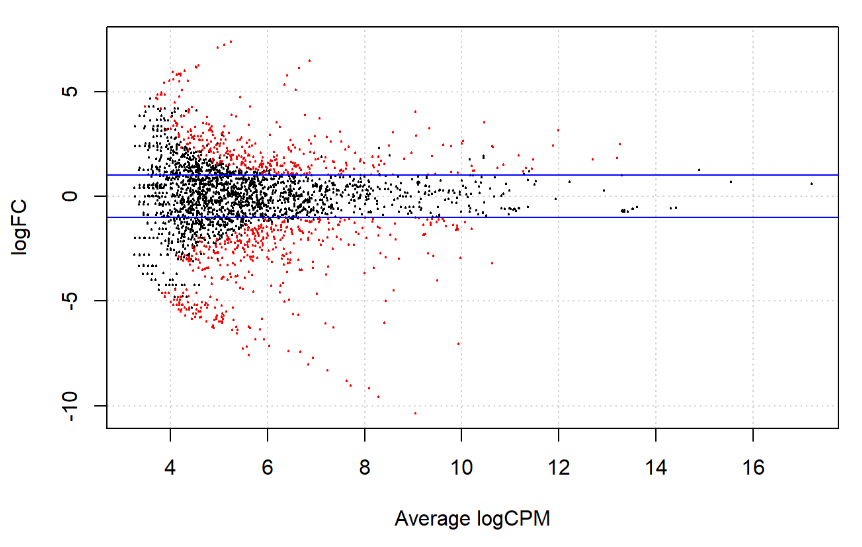
图中红色的是统计学上的显著差异表达基因,横轴的Average logCPM是校正后的样本间logCPM的均值
至于edgeR与DESeq2的比较其实已经有很多benchmark的文献做过了,这里就先鸽一下,以后有机会再来填坑。
benchmark ref:https://bioconductor.org/packages/release/bioc/vignettes/SummarizedBenchmark/inst/doc/Feature-Iterative.html
参考文章:
完。