通常,在计算TPM或RPKM/FPKM等基因表达量时,除了基因的counts信息外,我们还需要知道基因的长度。这里所用到的基因长度并不是某个基因在基因组上的完整长度。在基因表达分析中,“基因长度”通常指的是成熟转录本的长度,也就是无内含子的碱基序列。因此,单纯地使用基因的染色体起始和结束坐标相减并不能返回转录本的长度信息。目前,对于基因长度有多种定义,包括:
1. 基因最长转录本;
2. 多个转录本长度的平均值;
3. 非重叠外显子长度之和
4. 非重叠CDS序列长度之和
本文介绍使用gtf文件在R中获取基因长度(非重叠外显子长度之和)的方法
基因长度的定义
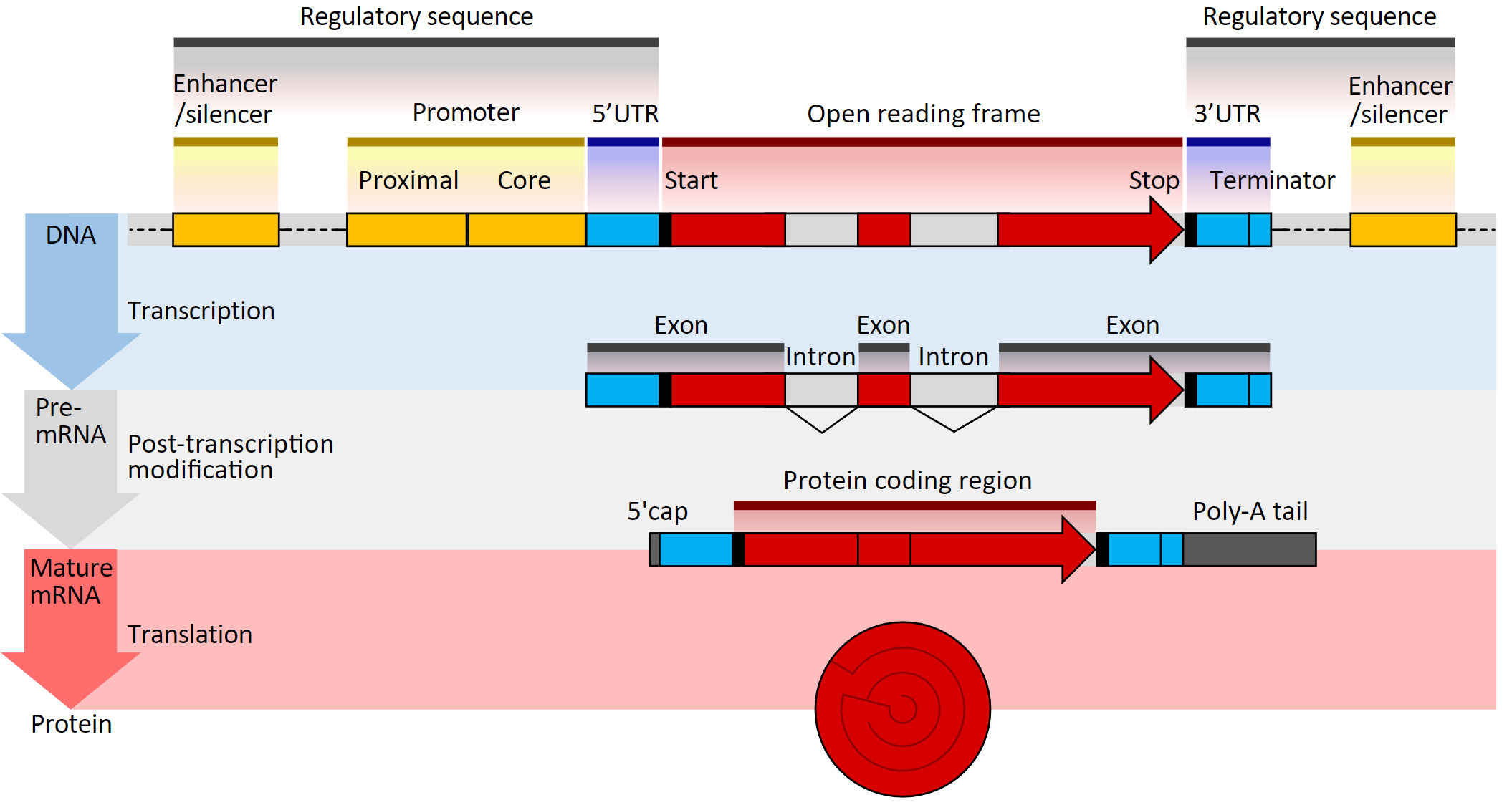
Figure Source: Gene structure
在真核生物,一个基因的DNA序列主要可以分为调控元件和编码序列,而编码序列又可以进一步分为外显子(Exon)和内含子(Intron)序列。经过剪接后,成熟的mRNA一般不包含内含子序列。所以,在统计基因长度时,也只考虑外显子的长度。由于可变剪接的存在,一个基因可能会有多个转录本,在进行基因水平的表达分析时,我们并不会区分各个转录本剪接变体的表达量,而是以基因为单位进行统计。因此,通过合并重叠外显子的区域来获得每个基因的非重叠外显子长度是一种基因长度估算的方法。
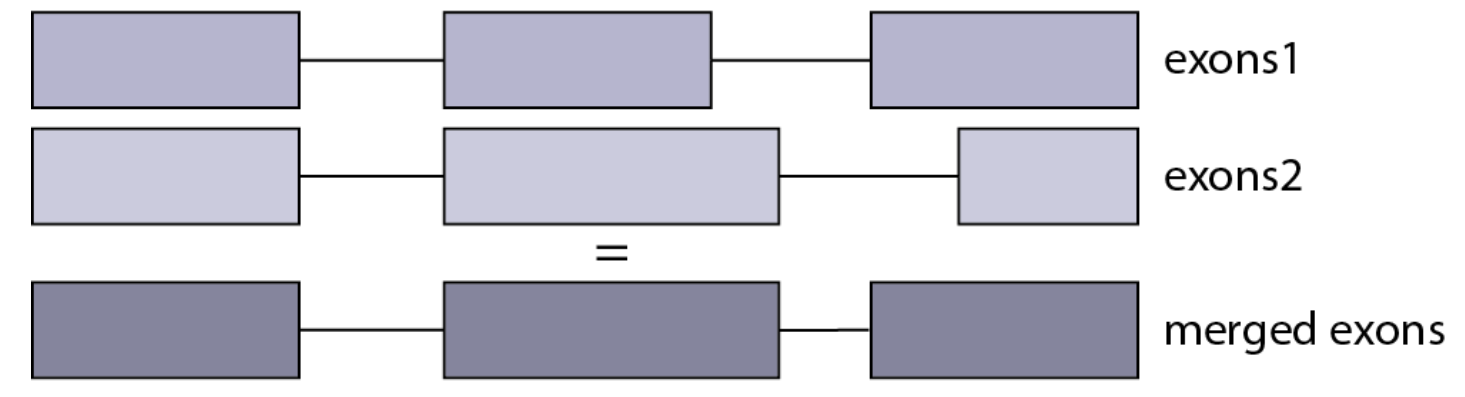
Figure Source: Schematic of non-overlapping exons
R实现 – 从GTF文件中获取基因长度
- 首先,读取计算基因counts时用的GTF文件,并将其转换为
TxDb对象; - 然后,提取每个基因的外显子注释信息;
- 最后,合并重叠的外显子,计算非重叠外显子的长度作为基因长度(bp)
1 | # Non-Overlapping Exon Length ---- |
在使用featureCounts统计基因counts时,其输出的counts.txt文件中通常会包含一列长度信息Length
我们可以比较一下使用相同注释文件时,两个程序计算的基因长度是否一致。
查看基因ENSMUSG00000000001.4:
1 | $ awk 'NR==2 || /ENSMUSG00000000001.4/' counts.txt |
可以发现featureCoutns输出的基因长度与我们计算的一致,这是由于featureCounts也是采用非重叠外显子作为基因长度。
featureCountsDocumentation:“For each meta-feature, the Length column gives the total length of genomic regions covered by features included in that meta-feature. Note that this length will be less than the sum of lengths of features included in the meta-feature when there are features overlapping with each other.”
以上就是在R中获取基因长度的方法。
Ref:
Tutorial: Extract Total Non-Overlapping Exon Length Per Gene With Bioconductor:
https://www.biostars.org/p/83901/How featureCounts define the gene length: https://support.bioconductor.org/p/88133/
完。