Geneformer for cell type annotation
Geneformer 是一个基于30M scRNA-seq data训练的Transformer模型,该训练数据包括人类的多种组织器官。Geneformer可以用于细胞水平的分类预测和基因水平的分类预测(例如预测是否为耐药基因),这里我们先根据教程演示其在细胞类型预测上的步骤。
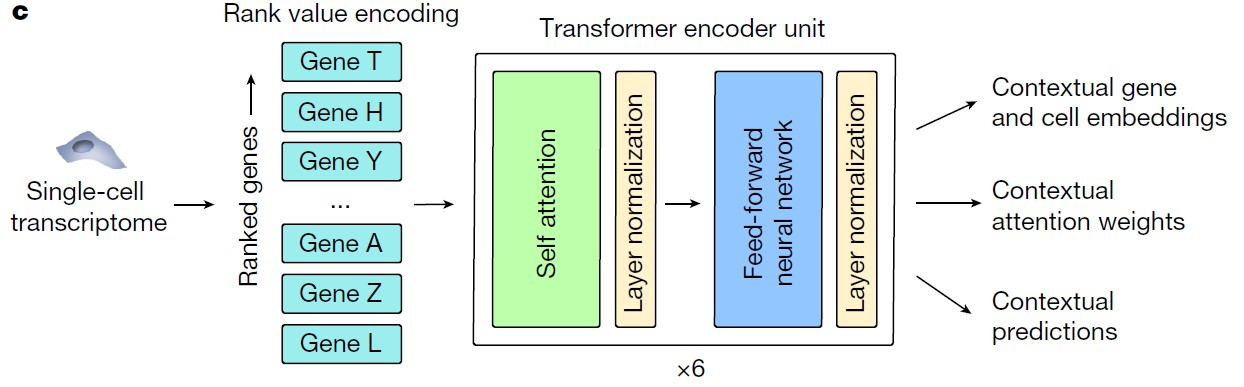
Geneformer首先将细胞的基因表达量转换为rank value encoding作为输入,再传递到transformer架构中进行预训练,后续可以根据下游任务在特定数据集上微调加上最后的输出层。
Geneformer 和其训练数据集 Genecorpus-30M 都可以在hugging face上访问到。
Geneformer环境配置
我们首先配置Geneformer分析所需要的环境。
创建conda环境
1 | conda create --envs geneformer |
为了在jupyter notebook使用该环境,我们需要安装ipykernel.
https://blog.csdn.net/mighty13/article/details/119859242
2
python -m ipykernel install --user --name geneformer
Installation
接着,我们下载Geneformer和相关的示例数据集。
Clone project
1 | git clone https://huggingface.co/ctheodoris/Geneformer |
Download associated dataset
1 | git clone https://huggingface.co/datasets/ctheodoris/Genecorpus-30M |
由于clone的.dataset相关文件只有1kb,我们需要手动下载相应训练数据
1 | cd Genecorpus-30M/example_input_files/cell_classification/cell_type_annotation/cell_type_train_data.dataset |
Install related modules
1 | pip3 install seaborn |
Import modules
1 | import os |
1 | # imports |
Prepare training and evaluation datasets
1 | # load cell type dataset (includes all tissues) |
我们读入文章提供的数据集Genecorpus-30M,该数据集以Apache Arrow format提供。
Data Fields
- cell_type
- organ_major
- input_id: rank value encoding for an example cell
- length: length of rank value encoding for that example cell
For rank value
- 计算各个检测到的基因在所有细胞中的非零中位值(nonzero median);
- 对每个细胞中的基因read counts除以该细胞的总read counts以校正测序深度;
- 对每个细胞的每个基因除以其相应的非零中位值以求得normalized expression;
- 基于每个细胞的normalized expression进行ranking,获得rank values。
The rank value encodings for each single cell transcriptome were then tokenized based on a total vocabulary of 25,424 protein-coding or miRNA genes detected within Geneformer-30M. The token dictionary mapping each token ID to special tokens (pad and mask) or Ensembl IDs for each gene is included within the repository as a pickle file (token_dictionary.pkl).
Why the rank values do not range from 1 to the number of genes in that cell?
1 | # elements of train_dataset |
Dataset({
features: ['cell_type', 'input_ids', 'length', 'organ_major'],
num_rows: 249556
})
Counter({'B cell (Plasmocyte)': 20728, 'T cell': 16695, 'Enterocyte progenitor': 15441, 'Fetal epithelial progenitor': 14580, 'Fetal neuron': 12287, 'Fetal mesenchymal progenitor': 11905, 'Erythroid progenitor cell (RP high)': 10819, 'Hepatocyte/Endodermal cell': 9781, 'Fetal enterocyte ': 9613, 'Erythroid cell': 9089, 'Loop of Henle': 8439, 'Macrophage': 7854, 'Monocyte': 7541, 'Epithelial cell': 7458, 'AT2 cell': 7333, 'Neutrophil': 7276, 'Fibroblast': 6980, 'Dendritic cell': 5960, 'Pancreas exocrine cell': 5538, 'Endothelial cell (APC)': 5431, 'M2 Macrophage': 5373, 'Endothelial cell': 4738, 'Antigen presenting cell (RPS high)': 4658, 'Intercalated cell': 4414, 'Sinusoidal endothelial cell': 3844, 'B cell': 3783, 'Endothelial cell (endothelial to mesenchymal transition)': 3647, 'Fetal acinar cell': 3214, 'Ureteric bud cell': 2472, 'Enterocyte': 1994, 'Proximal tubule progenitor': 1846, 'Smooth muscle cell': 1794, 'Fetal stromal cell': 1061, 'Stromal cell': 1029, 'Mast cell': 968, 'Fetal endocrine cell': 834, 'Neutrophil (RPS high)': 604, 'Intermediated cell': 529, 'Proliferating T cell': 528, 'CB CD34+': 423, 'Basal cell': 236, 'Primordial germ cell': 230, 'Fetal fibroblast': 125, 'Fetal Neuron': 114, 'Stratified epithelial cell': 113, 'Fetal skeletal muscle cell': 57, 'Fetal chondrocyte': 49, 'Mesothelial cell': 30, 'Goblet cell': 19, 'Chondrocyte': 19, 'hESC': 18, 'Fasciculata cell': 13, 'Gastric endocrine cell': 7, 'Myeloid cell': 7, 'Epithelial cell (intermediated)': 7, 'Astrocyte': 4, 'Kidney intercalated cell': 3, 'Ventricle cardiomyocyte': 2, 'Immature sertoli cell (Pre-Sertoli cell)': 2})
Counter({'large_intestine': 50363, 'kidney': 45059, 'lung': 33309, 'liver': 28376, 'pancreas': 28116, 'immune': 17110, 'spleen': 15614, 'brain': 13440, 'placenta': 9509, 'bone_marrow': 8660})
569
569
1 | print(train_dataset['length'][2]) |
625
9
25414
接着,我们将每个组织的细胞分为80%的training set和20%的evaluation set以供后续的model fine-tune使用。
这里的细胞都带有celltype labels,并转换为数值标记,例如”B cell” – 1。之后提供给下游fine-tune训练使用。
1 | dataset_list = [] |
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-d895a8dc1f433c21_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-5f89f392ef5206d4_*_of_00006.arrow
Loading cached shuffled indices for dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-ed12a33637a220d0.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-37f89eeea757d6c5_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-74f78f156da669e5_*_of_00004.arrow
spleen
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-f6dc5b7fe424bf2d_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-896e8ac5f576851c_*_of_00006.arrow
Loading cached shuffled indices for dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-820cdcb7f7383e21.arrow
kidney
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-d4006d9701718093_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-3d777eac360cb136_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-0292fb0af10803a4_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-a996d2bf76adce7c_*_of_00006.arrow
Loading cached shuffled indices for dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-65960d687cc54b82.arrow
lung
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-456daf9851000084_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-60719877e54cdb77_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-5304f89297ce82b0_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-5284c824687e6edd_*_of_00006.arrow
Loading cached shuffled indices for dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-01ed43e584533226.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-53c0460a585f1ee6_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-3d719e36692d0047_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-f94573c555aec2c1_*_of_00004.arrow
brain
placenta
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-c546b4750f90df75_*_of_00006.arrow
Loading cached shuffled indices for dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-f29113e9ecf5a9a6.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-3e7c6c00c9efd043_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-f57326d9b39be686_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-d155ab4f91b9b109_*_of_00004.arrow
immune
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-7699e47999d0e20a_*_of_00006.arrow
Loading cached shuffled indices for dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-c7fa1566fa301d6f.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-c30b7a2b73e574a5_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-b2e2f437bfe2e62b_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-f30bce417351df8c_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-f040851bda405e47_*_of_00006.arrow
large_intestine
Loading cached shuffled indices for dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-fc4569333911ef13.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-791285ddf886e3a6_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-06ee2b3139e1b0a8_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-2ae398bc8c532f07_*_of_00004.arrow
pancreas
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-a47c5e32577914f3_*_of_00006.arrow
Loading cached shuffled indices for dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-0bc8012540760dc1.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-d4dcf029b0b1437a_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-f2a3bf8b55c9560b_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-8434ffd865a76d79_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-dbd87150b1b95134_*_of_00006.arrow
Loading cached shuffled indices for dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-b56b8ddf9ca9920d.arrow
liver
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-a31834f681c29f0f_*_of_00004.arrow
Loading cached processed dataset at D:\jupyterNote\Geneformer\Genecorpus-30M\example_input_files\cell_classification\cell_type_annotation\cell_type_train_data.dataset\cache-c65ae92f453eb94b_*_of_00004.arrow
每个组织的celltype和label id的映射关系存储在target_dict_list这个list中
1 | # number of cells for each organ |
spleen
Counter({'Endothelial cell (APC)': 5, 'Macrophage': 4, 'Neutrophil': 3, 'T cell': 2, 'B cell': 1, 'B cell (Plasmocyte)': 0})
kidney
Counter({'T cell': 14, 'Dendritic cell': 13, 'Fetal stromal cell': 12, 'Smooth muscle cell': 11, 'Endothelial cell': 10, 'Macrophage': 9, 'Proximal tubule progenitor': 8, 'Intermediated cell': 7, 'Fetal mesenchymal progenitor': 6, 'Intercalated cell': 5, 'Ureteric bud cell': 4, 'Loop of Henle': 3, 'Fetal epithelial progenitor': 2, 'Epithelial cell': 1, 'Endothelial cell (APC)': 0})
lung
Counter({'Basal cell': 15, 'Monocyte': 14, 'Endothelial cell': 13, 'Proliferating T cell': 12, 'Dendritic cell': 11, 'Fetal epithelial progenitor': 10, 'B cell (Plasmocyte)': 9, 'Mast cell': 8, 'Endothelial cell (APC)': 7, 'T cell': 6, 'Endothelial cell (endothelial to mesenchymal transition)': 5, 'M2 Macrophage': 4, 'Macrophage': 3, 'Fetal mesenchymal progenitor': 2, 'AT2 cell': 1, 'Smooth muscle cell': 0})
brain
Counter({'Fetal epithelial progenitor': 5, 'Fetal endocrine cell': 4, 'Erythroid cell': 3, 'Macrophage': 2, 'Fetal mesenchymal progenitor': 1, 'Fetal neuron': 0})
placenta
Counter({'Macrophage': 2, 'Epithelial cell': 1, 'Fibroblast': 0})
immune
Counter({'B cell': 9, 'Neutrophil (RPS high)': 8, 'B cell (Plasmocyte)': 7, 'Dendritic cell': 6, 'Erythroid progenitor cell (RP high)': 5, 'Erythroid cell': 4, 'Neutrophil': 3, 'T cell': 2, 'Monocyte': 1, 'Antigen presenting cell (RPS high)': 0})
large_intestine
Counter({'Endothelial cell': 15, 'Smooth muscle cell': 14, 'Fetal stromal cell': 13, 'B cell': 12, 'Stromal cell': 11, 'Enterocyte': 10, 'T cell': 9, 'Macrophage': 8, 'Dendritic cell': 7, 'Epithelial cell': 6, 'Fetal neuron': 5, 'Fetal mesenchymal progenitor': 4, 'B cell (Plasmocyte)': 3, 'Fetal enterocyte ': 2, 'Hepatocyte/Endodermal cell': 1, 'Enterocyte progenitor': 0})
pancreas
Counter({'Endothelial cell (APC)': 14, 'Smooth muscle cell': 13, 'Dendritic cell': 12, 'Fetal epithelial progenitor': 11, 'Erythroid cell': 10, 'Fetal endocrine cell': 9, 'Endothelial cell': 8, 'Enterocyte progenitor': 7, 'Fetal neuron': 6, 'Macrophage': 5, 'Fetal mesenchymal progenitor': 4, 'Pancreas exocrine cell': 3, 'Fetal acinar cell': 2, 'T cell': 1, 'B cell': 0})
liver
Counter({'CB CD34+': 11, 'B cell': 10, 'Neutrophil (RPS high)': 9, 'B cell (Plasmocyte)': 8, 'T cell': 7, 'Neutrophil': 6, 'Monocyte': 5, 'Dendritic cell': 4, 'Macrophage': 3, 'Sinusoidal endothelial cell': 2, 'Erythroid cell': 1, 'Erythroid progenitor cell (RP high)': 0})
1 | trainset_dict = dict(zip(organ_list, dataset_list)) |
Fine-Tune With Cell Classification Learning Objective and Quantify Predictive Performance
接下来,使用预设的hyperparameters进行训练,作者建议根据下游任务调整hyperparameters。
另外,我们定义一个评估模型预测性能的函数compute_metrics.
1 | def compute_metrics(pred): |
1 | # set model parameters |
接着,我们对每个组织都微调一个细胞分类的预测器,其中包括, brain, immune, kidney, large intestine, liver, lung, pancreas, placenta, and spleen.
这一步耗时很久
模型通过BertForSequenceClassification.from_pretrained读入。num_labels为模型输出的class数目,这里设置成每个组织对应的细胞类型数量即可。
随后,创建trainer并进行训练,.predict()进行预测。
1 | for organ in organ_list: |
1 | Some weights of the model checkpoint at D:\jupyterNote\Geneformer were not used when initializing BertForSequenceClassification: ['cls.predictions.transform.dense.weight', 'cls.predictions.transform.LayerNorm.weight', 'cls.predictions.decoder.bias', 'cls.predictions.transform.LayerNorm.bias', 'cls.predictions.bias', 'cls.predictions.decoder.weight', 'cls.predictions.transform.dense.bias'] |
训练结束后,训练的模型,及其预测结果都输出到设置的output_dir下。
每个文件夹中都包含了fine-tuned model相关文件(training_args.bin, config.json, pytorch_model.bin),以及预测结果相关文件(predictions.pickle)
1 | $ ls 230719_geneformer_CellClassifier_brain_L2048_B4_LR5e-05_LSlinear_WU500_E10_Oadamw_F0/ |
1 | # clear GPU memory after pytorch training |
1 | # The pretrained model |
BertForSequenceClassification(
(bert): BertModel(
(embeddings): BertEmbeddings(
(word_embeddings): Embedding(25426, 256, padding_idx=0)
(position_embeddings): Embedding(2048, 256)
(token_type_embeddings): Embedding(2, 256)
(LayerNorm): LayerNorm((256,), eps=1e-12, elementwise_affine=True)
(dropout): Dropout(p=0.02, inplace=False)
)
(encoder): BertEncoder(
(layer): ModuleList(
(0-5): 6 x BertLayer(
(attention): BertAttention(
(self): BertSelfAttention(
(query): Linear(in_features=256, out_features=256, bias=True)
(key): Linear(in_features=256, out_features=256, bias=True)
(value): Linear(in_features=256, out_features=256, bias=True)
(dropout): Dropout(p=0.02, inplace=False)
)
(output): BertSelfOutput(
(dense): Linear(in_features=256, out_features=256, bias=True)
(LayerNorm): LayerNorm((256,), eps=1e-12, elementwise_affine=True)
(dropout): Dropout(p=0.02, inplace=False)
)
)
(intermediate): BertIntermediate(
(dense): Linear(in_features=256, out_features=512, bias=True)
(intermediate_act_fn): ReLU()
)
(output): BertOutput(
(dense): Linear(in_features=512, out_features=256, bias=True)
(LayerNorm): LayerNorm((256,), eps=1e-12, elementwise_affine=True)
(dropout): Dropout(p=0.02, inplace=False)
)
)
)
)
(pooler): BertPooler(
(dense): Linear(in_features=256, out_features=256, bias=True)
(activation): Tanh()
)
)
(dropout): Dropout(p=0.02, inplace=False)
(classifier): Linear(in_features=256, out_features=12, bias=True)
)
接下来,我们将基于immune数据的微调模型应用到3k PBMCs scRNA-seq data上进行celltype prediction.
首先,我们需要将原始的测序counts转换为rank values encoding (tk.tokenize_data).
1 | # applying fine-tuned model on new datasets |
Tokenizing D:\jupyterNote\pySC\output\pbmc3k.loom
D:\jupyterNote\pySC\output\pbmc3k.loom has no column attribute 'filter_pass'; tokenizing all cells.
读入tokenized dataset
1 | # 2. load new dataset |
Dataset({
features: ['input_ids', 'cell_type', 'organ_major', 'length'],
num_rows: 2638
})
1 | import pandas as pd |
| input_ids | cell_type | organ_major | length | |
|---|---|---|---|---|
| 0 | [19693, 10551, 2362, 1869, 4658, 18585, 9039, ... | CD4 T | immune | 139 |
| 1 | [5307, 10632, 8073, 3539, 19629, 516, 18552, 1... | B | immune | 247 |
| 2 | [6729, 226, 9004, 11666, 4621, 1433, 8198, 327... | CD4 T | immune | 200 |
| 3 | [3057, 12015, 17654, 6179, 2522, 2770, 417, 62... | FCGR3A Monocytes | immune | 185 |
| 4 | [12623, 18649, 10321, 2313, 7245, 13219, 242, ... | NK | immune | 91 |
| ... | ... | ... | ... | ... |
| 2633 | [2522, 13139, 3539, 449, 19629, 488, 2770, 695... | CD14 Monocytes | immune | 241 |
| 2634 | [13492, 6556, 12734, 3078, 3539, 643, 695, 195... | B | immune | 189 |
| 2635 | [14848, 16634, 19629, 2987, 5214, 11314, 17576... | B | immune | 108 |
| 2636 | [19629, 19899, 14408, 3078, 7380, 1868, 4472, ... | B | immune | 93 |
| 2637 | [4658, 18585, 18828, 14377, 10632, 13116, 1481... | CD4 T | immune | 118 |
2638 rows × 4 columns
由于模型要求input tensors(每个细胞的rank encoding)长度一致,这里将其padding到统一长度(所有细胞中最多的基因数)。
1 | from geneformer.pretrainer import token_dictionary |
1 | # padded to be the same length. |
Loading cached processed dataset at D:\jupyterNote\Geneformer\examples\token_data\tk_pbmc3k.dataset\cache-4214517ba3d677b2.arrow
1 | pd.DataFrame(padded_dataset) |
| input_ids | cell_type | organ_major | length | attention_mask | |
|---|---|---|---|---|---|
| 0 | [19693, 10551, 2362, 1869, 4658, 18585, 9039, ... | CD4 T | immune | 139 | [1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, ... |
| 1 | [5307, 10632, 8073, 3539, 19629, 516, 18552, 1... | B | immune | 247 | [1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, ... |
| 2 | [6729, 226, 9004, 11666, 4621, 1433, 8198, 327... | CD4 T | immune | 200 | [1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, ... |
| 3 | [3057, 12015, 17654, 6179, 2522, 2770, 417, 62... | FCGR3A Monocytes | immune | 185 | [1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, ... |
| 4 | [12623, 18649, 10321, 2313, 7245, 13219, 242, ... | NK | immune | 91 | [1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, ... |
| ... | ... | ... | ... | ... | ... |
| 2633 | [2522, 13139, 3539, 449, 19629, 488, 2770, 695... | CD14 Monocytes | immune | 241 | [1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, ... |
| 2634 | [13492, 6556, 12734, 3078, 3539, 643, 695, 195... | B | immune | 189 | [1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, ... |
| 2635 | [14848, 16634, 19629, 2987, 5214, 11314, 17576... | B | immune | 108 | [1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, ... |
| 2636 | [19629, 19899, 14408, 3078, 7380, 1868, 4472, ... | B | immune | 93 | [1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, ... |
| 2637 | [4658, 18585, 18828, 14377, 10632, 13116, 1481... | CD4 T | immune | 118 | [1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, ... |
2638 rows × 5 columns
接下来,读入微调模型进行预测
1 | # 3. load the fine-tuned model |
1 | BertForSequenceClassification( |
使用fine-tuned model进行分类预测,这里根据最大预测值判断cell type。
1 | # celltype : index |
2638
1 | print(ct_pred_id[0:10]) |
[2 9 2 1 2 9 2 2 2 1]
['T cell', 'B cell', 'T cell', 'Monocyte', 'T cell', 'B cell', 'T cell', 'T cell', 'T cell', 'Monocyte']
{'Antigen presenting cell (RPS high)': 0, 'Monocyte': 1, 'T cell': 2, 'Neutrophil': 3, 'Erythroid cell': 4, 'Erythroid progenitor cell (RP high)': 5, 'Dendritic cell': 6, 'B cell (Plasmocyte)': 7, 'Neutrophil (RPS high)': 8, 'B cell': 9}
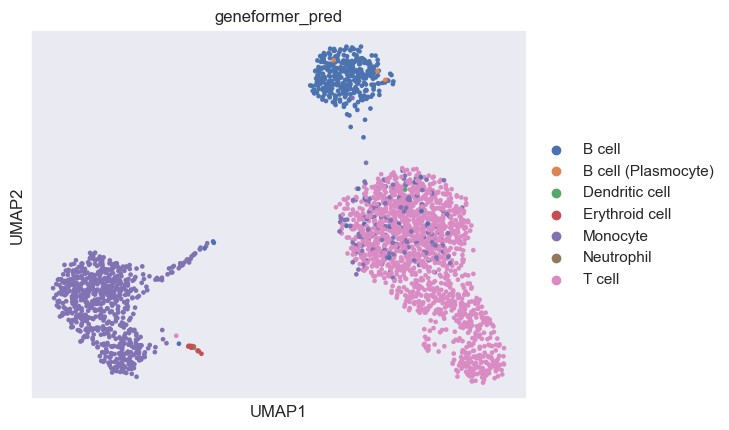
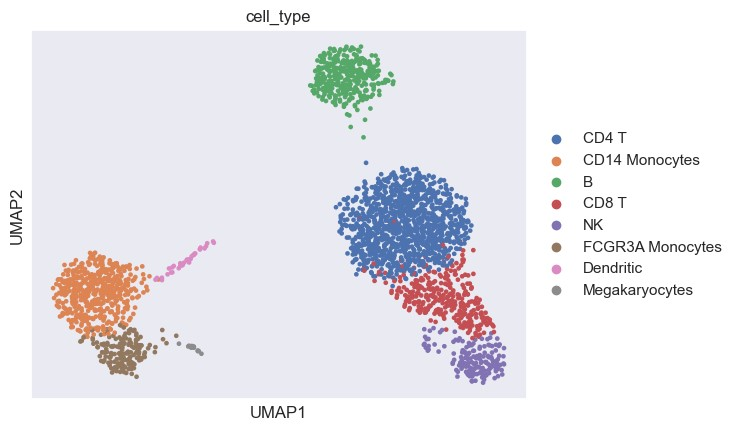
用UMAP可视化细胞分类的结果
1 | import numpy as np |
AnnData object with n_obs × n_vars = 2638 × 1838
obs: 'n_genes', 'n_genes_by_counts', 'n_counts', 'total_counts_mt', 'pct_counts_mt', 'leiden', 'cell_type', 'organ_major'
var: 'ensembl_id', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std', 'gene_name'
uns: 'hvg', 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'rank_genes_groups', 'umap'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts', 'data', 'scaled'
obsp: 'connectivities', 'distances'
尽管Geneformer对细胞的名称和原数据不太一样,我们可以看到Geneformer注释的结果大体上和原本注释是一致的。总的来说,Geneformer可以作为一种细胞类型预测的工具使用,但最好先对预训练模型微调,这要求我们有相关的单细胞数据集进行微调训练。
1 | adata.obs['geneformer_pred'] = ct_pred_label |
E:\miniconda3\envs\geneformer\lib\site-packages\scanpy\plotting\_tools\scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
E:\miniconda3\envs\geneformer\lib\site-packages\scanpy\plotting\_tools\scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
1 | adata.obs |
| n_genes | n_genes_by_counts | n_counts | total_counts_mt | pct_counts_mt | leiden | cell_type | organ_major | |
|---|---|---|---|---|---|---|---|---|
| AAACATACAACCAC-1 | 781 | 779 | 2419.0 | 73.0 | 3.017776 | CD4 T | CD4 T | immune |
| AAACATTGAGCTAC-1 | 1352 | 1352 | 4903.0 | 186.0 | 3.793596 | B | B | immune |
| AAACATTGATCAGC-1 | 1131 | 1129 | 3147.0 | 28.0 | 0.889736 | CD4 T | CD4 T | immune |
| AAACCGTGCTTCCG-1 | 960 | 960 | 2639.0 | 46.0 | 1.743085 | FCGR3A Monocytes | FCGR3A Monocytes | immune |
| AAACCGTGTATGCG-1 | 522 | 521 | 980.0 | 12.0 | 1.224490 | NK | NK | immune |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
| TTTCGAACTCTCAT-1 | 1155 | 1153 | 3459.0 | 73.0 | 2.110436 | CD14 Monocytes | CD14 Monocytes | immune |
| TTTCTACTGAGGCA-1 | 1227 | 1224 | 3443.0 | 32.0 | 0.929422 | B | B | immune |
| TTTCTACTTCCTCG-1 | 622 | 622 | 1684.0 | 37.0 | 2.197150 | B | B | immune |
| TTTGCATGAGAGGC-1 | 454 | 452 | 1022.0 | 21.0 | 2.054795 | B | B | immune |
| TTTGCATGCCTCAC-1 | 724 | 723 | 1984.0 | 16.0 | 0.806452 | CD4 T | CD4 T | immune |
2638 rows × 8 columns
总结
对于细胞分类的微调,我们需要:
- 获取组织对应的微调数据集,并且有细胞的label信息,例如各个细胞类型;
关于数据集大小,从作者提供的例子来看,最少的情况是884个细胞,但其余下游任务都超过10k细胞
- 以
BertForSequenceClassification的方式读入预训练模型,并设置num_labels为分类数目; - 根据微调的数据集训练,加上最后的输出层(task-specific transformer layer),并对微调模型预测性能进行评估;
- 在新的数据集上应用微调模型进行预测。
Ref:
Transfer learning enables predictions in network biology: https://doi.org/10.1038/s41586-023-06139-9