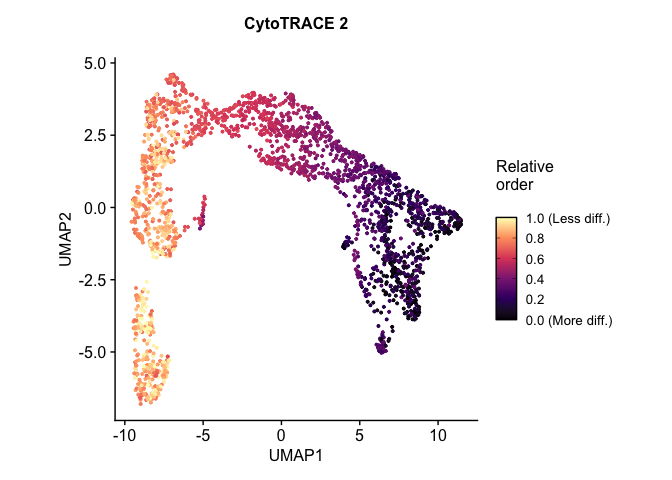
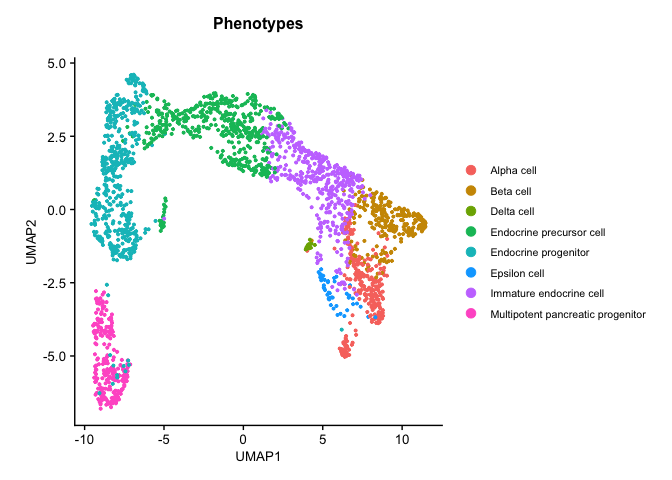
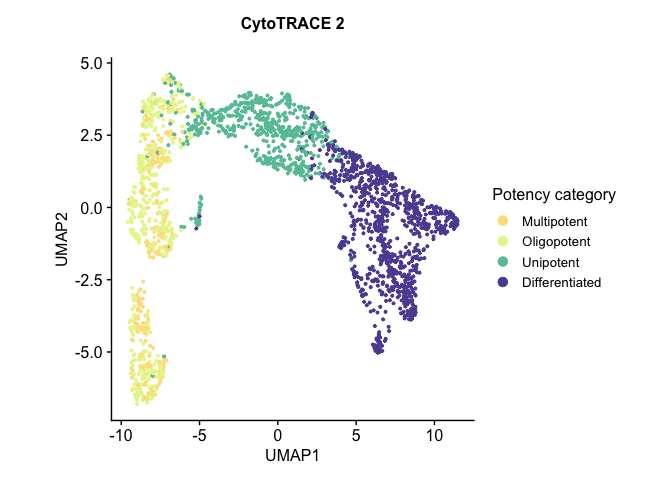
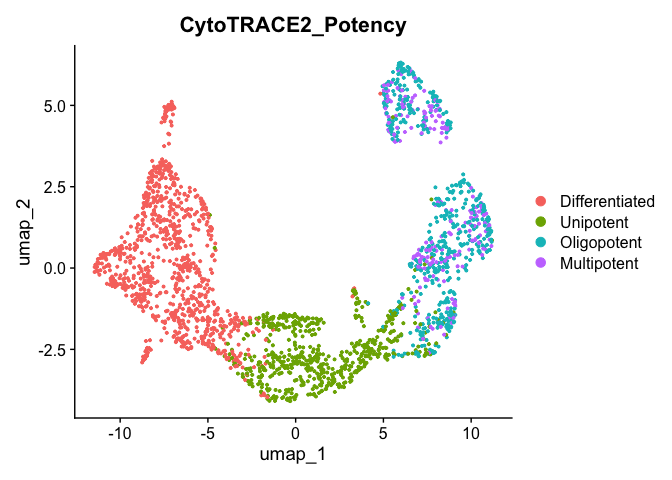
# download the .rds file (this will download the file to your working directory) download.file("https://drive.google.com/uc?export=download&id=1ivi9TBlmzVTDGzNWQrXXeyL68Wug989K", "Pancreas_10x_downsampled.rds")
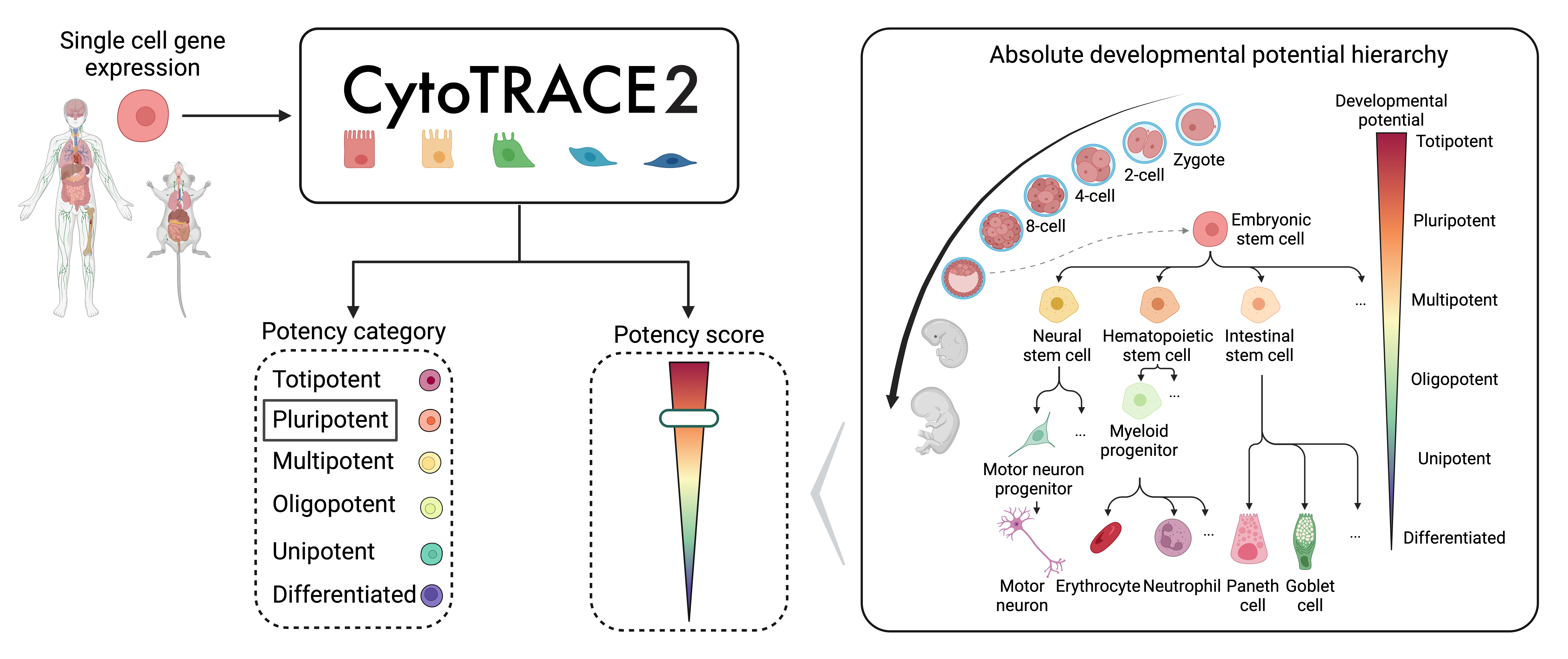
运行CytoTRACE2
运行 cytotrace2() 进行细胞干性推断,函数运行时执行以下操作:
预处理数据:在每个细胞中对表达数据进行降序排序,使用排序后的秩(rank)进行预测
预测细胞发育状态:读入模型预训练参数进行预测
1 2 3 4
# full model parameter_dict <- readRDS(system.file("extdata", "parameter_dict_17.rds", package = "CytoTRACE2")) # else parameter_dict <- readRDS(system.file("extdata", "parameter_dict_5_best.rds", package = "CytoTRACE2"))
## You have loaded plyr after dplyr - this is likely to cause problems. ## If you need functions from both plyr and dplyr, please load plyr first, then dplyr: ## library(plyr); library(dplyr)
## The following object is masked from 'package:dplyr': ## ## nth
## The following object is masked from 'package:data.table': ## ## transpose
## Loading required package: Seurat
## Loading required package: SeuratObject
## Loading required package: sp
## 'SeuratObject' was built under R 4.4.0 but the current version is ## 4.4.1; it is recomended that you reinstall 'SeuratObject' as the ABI ## for R may have changed
## ## Attaching package: 'SeuratObject'
## The following objects are masked from 'package:base': ## ## intersect, t
## Loading required package: stringr
## Warning: replacing previous import 'data.table::first' by 'dplyr::first' when ## loading 'CytoTRACE2'
## Warning: replacing previous import 'data.table::last' by 'dplyr::last' when ## loading 'CytoTRACE2'
## Warning: replacing previous import 'data.table::between' by 'dplyr::between' ## when loading 'CytoTRACE2'
library(Seurat) seu <- CreateSeuratObject(expression_data, meta.data = annotation)
## Warning: Data is of class data.frame. Coercing to dgCMatrix.
seu
## An object of class Seurat ## 17326 features across 2280 samples within 1 assay ## Active assay: RNA (17326 features, 0 variable features) ## 1 layer present: counts
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck ## ## Number of nodes: 2280 ## Number of edges: 73179 ## ## Running Louvain algorithm... ## Maximum modularity in 10 random starts: 0.9505 ## Number of communities: 4 ## Elapsed time: 0 seconds
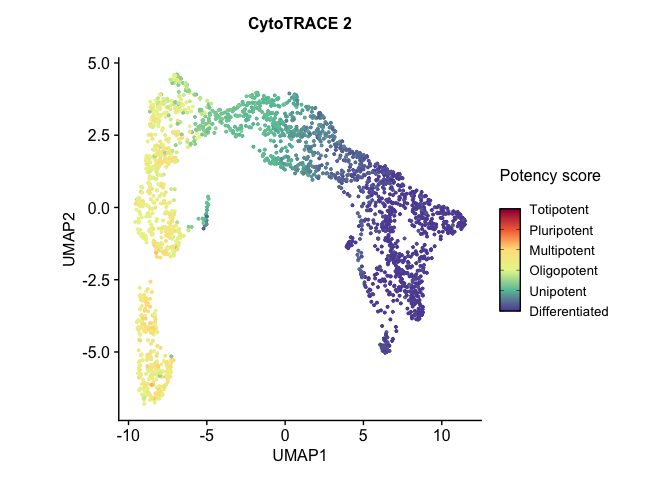
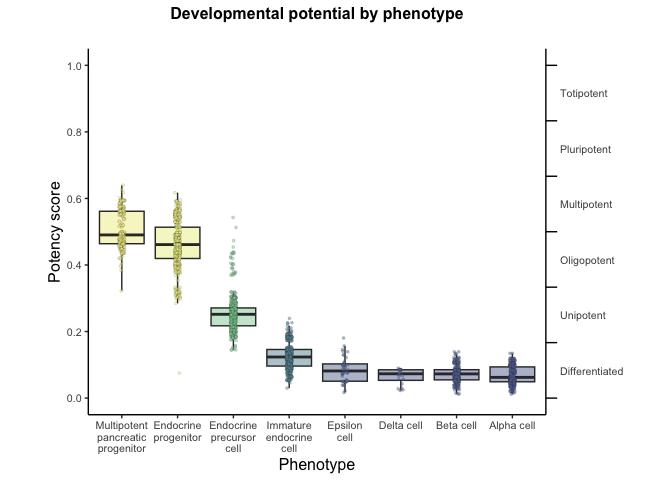
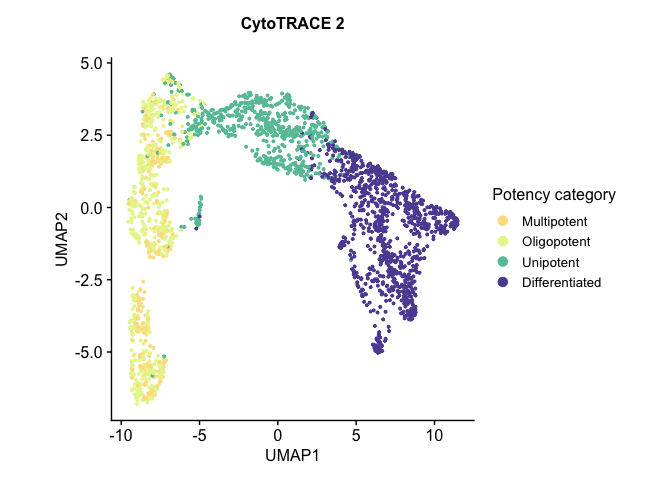
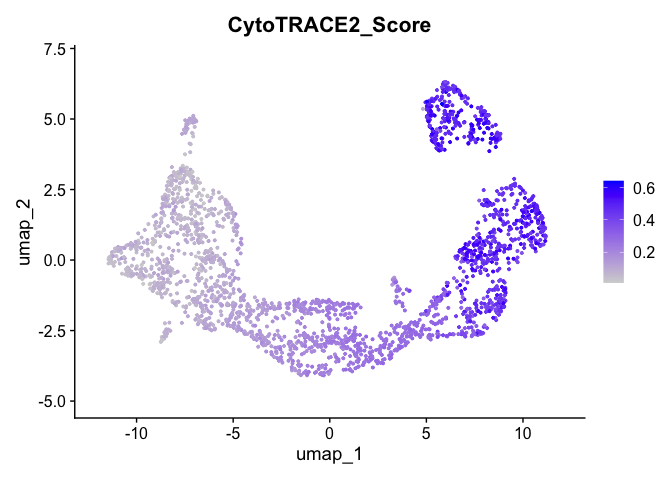
# Run UMAP cytotrace2_result <- RunUMAP(cytotrace2_result, dims = 1:20)
## 14:12:35 UMAP embedding parameters a = 0.9922 b = 1.112
## 14:12:35 Read 2280 rows and found 20 numeric columns
## 14:12:35 Using Annoy for neighbor search, n_neighbors = 30
## 14:12:35 Building Annoy index with metric = cosine, n_trees = 50